Artikel
Josivan Gomes Lima1*, Marcel Catão Ferreira dos Santos1, Julliane Tamara Araújo de Melo Campos2
1Departamento de medicina clínica, disciplina de endocrinologia e metabologia. Krankenhaus Universitário Onofre Lopes, Universidade Federal do Rio Grande do Norte (UFRN), Natal, RN, Brasilien
2Faculty of Health Sciences of Trairi, Federal University of Rio Grande do North (UFRN), Natal, RN, Brasilien
Zusammenfassung
Die angeborene generalisierte Lipodystrophie (CGL) ist eine seltene und schwere autosomale rezessive Krankheit. Patienten sind in der Speicherung von Körperfett defekt und lagern folglich Fett in ektopen Geweben, hauptsächlich in der Leber, ab und können eine Zirrhose entwickeln. Insulinresistenz ist ein typischer Befund, der Diabetes verursacht, der hohe tägliche Insulindosen erfordert. Im Bundesstaat Rio Grande do Norte, Brasilien, haben wir eine der größten Kohorten von Patienten mit CGL. In diesem Artikel überprüfen wir die Pathophysiologie, das klinische Bild und die Behandlung dieser Krankheit.
Einleitung
Typ-2-Diabetes ist ein Weltgesundheitsproblem und resultiert normalerweise aus Übergewicht und erhöhtem viszeralem Fett, was zu einer peripheren Insulinresistenz und einer Unfähigkeit der Bauchspeicheldrüse führt, Insulin freizusetzen, um diese Resistenz auszugleichen. Andere weniger häufige Arten von Diabetes treten aufgrund spezifischer genetischer Mutationen auf, wie die angeborene generalisierte Lipodystrophie (CGL), auch bekannt als Berardinelli-Seip-angeborene Lipodystrophie (BSCL). CGL ist eine autosomal-rezessive Erkrankung, die basierend auf Genmutationen in vier Typen eingeteilt wird. Die veränderten Gene spielen wesentliche Funktionen für die Adipozytenbildung, die Lipidproduktion und die ordnungsgemäße Lagerung im Adipozyten. Die Mutationen verringern das Fettgewebe mit der daraus resultierenden Ablagerung von Fett an ektopischen Stellen, was zu Fettleber, verändertem Kohlenhydratstoffwechsel, schwerer Insulinresistenz mit Hyperinsulinämie und Akromegaloidmerkmalen und Dyslipidämie1-3 führt. Das CGL-Syndrom hat weltweit rund 500 Fälle gemeldet. In Brasilien, im Bundesstaat Rio Grande do Norte (RN), haben wir in den letzten 20 Jahren 54 Fälle diagnostiziert, behandelt und verfolgt4, 5. In einer deskriptiven Studie mit Sekundärdaten schätzten wir insgesamt 103 Patienten in RN6. Dies deutet auf eine viel höhere Prävalenz hin als in der Literatur berichtet (1: 1 million)7.
Triacylglycerinbildung und Speicherung in Lipidtröpfchen
Die Biosynthese von Triglyceriden und Phospholipiden (Abbildung 1A) beginnt mit der Glycerin-3-phosphat-Acyltransferase (GPAT), die das Glycerin-3-phosphat in Position 1 acyliert und 1-Acylglycerin-3-phosphat (Lysophosphatidsäure) bildet. Es folgt ein weiterer Acylierungsschritt an Position zwei durch das Enzym AGPAT (1-Acylglycerol-3-phosphat-acyltransferase), wobei 1,2-Diacylglycerol-3-phosphat (phosphatidsäure) entsteht. Es ist ein wichtiger Zwischenschritt im Biosyntheseweg von Triglyceriden und Phosphoglyceriden. Es gibt 11 Isoformen von AGPAT-Enzymen, die von verschiedenen Genen kodiert4. AGPAT1 und AGPAT2 sind die am ausführlichsten untersuchten. AGPAT1 ist in hohen Konzentrationen in Hoden, Bauchspeicheldrüse und in geringerem Maße im Fettgewebe und anderen Geweben wie Herz, Plazenta, Gehirn und Lunge vorhanden, während AGPAT2 im Fettgewebe reichlich vorhanden ist. In den folgenden Schritten bildet das cytosolische Enzym Phosphatidsäurephosphatase (PAP oder Lipin) 1,2-Diacylglycerin und die 1,2-Diacylglycerinacyltransferase (DGAT) bildet Triacylglycerol4. Phosphatidsäure und Diacylglycerin können auch andere Phospholipide wie Cardiolipin, Phosphatidylinositol und Phosphatidylcholin enthalten.
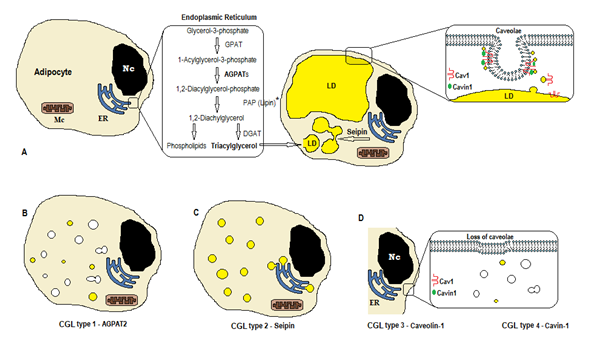
Abbildung 1. Schema der Triglyceridsynthese nach CGL-Typen. (A) Normale Synthese und Speicherung von Triacylglycerin (TAG) im Adipozyten. (B) Die Mutation von AGPAT2 verringert die TAG-Produktion (einige werden noch unter Stimulation anderer AGPATs synthetisiert). (C) Mutation des Seipin-Gens verringert die TAG-Synthese und die Bildung und Fusion von Lipidtröpfchen (LD). (D) Caveolin-1 und Cavin-1 werden zur Bildung und Stabilisierung der Caveolen benötigt. Eine Mutation in CAV1 (Typ 3) oder CAVIN1 (Typ 4) kann zum Verlust von Caveolen in der Membran führen. Nc, Kern. ER, endoplasmatisches Retikulum. Mc, Mitochondrien. * Lipin ist ein zytosolisches Enzym, das von Seipin in der ER verankert wird.
Diese Reaktionen treten im endoplasmatischen Retikulum (ER) der Adipozyten auf, wo eine fortschreitende Ansammlung von Triglyceriden die Bildung kleiner Lipidtröpfchen (LD) verursacht.8. Das Produkt des Gens BSCL2 ist ein Transmembranprotein namens Seipin, das die Fusion von kleinem LD mit großem LD verursacht. Seipin befindet sich im ER und konzentriert sich an der Verbindungsstelle mit dem entstehenden LD, was den Lipidverkehr zwischen ER und LD und den Einbau von Triglyceriden in LD9 erleichtert. Seipin kann auch als ER-Anker für das cytosolische Enzym Lipin1 wirken. Seipin ist nicht nur für die Lipidtröpfchenfusion, -größe und -morphologie notwendig, sondern auch für die Adipogenese (über Wechselwirkung mit Lipin 1) und die zelluläre Triglyceridlipolyse10, 11. Ein Mangel an Seipin behindert die Differenzierung von Prä-Adipozyten zu Adipozyten und beeinflusst die endgültige Reife9, wie Studien an mesenchymalen Stammzellen mit ausgeschlagenem BSCL2 zeigen12. Nicht-Fettgewebe exprimieren auch Seipin, und andere Funktionen sind zu bestimmen.
In den Adipozyten machen Caveolae, die spezialisierte 50-100nm-Membraninvaginationen sind, 20% der Plasmamembranfläche aus, wodurch die Adipozyten die Zellen mit der höchsten Dichte an Caveolae13 sind. Die Bildung von Lipidtröpfchen benötigt ein Membranprotein (Caveolin – der Hauptbestandteil von Caveolae-Membranen) und ein zytoplasmatisches Protein (Cavin-1)14. Die Gene CAV1, CAV2 und CAV3 kodieren für drei Formen von Caveolin mit ähnlichen Strukturen (Caveolin-1, Caveolin-2 und Caveolin-3). Caveolin-1 und Caveolin-2 sind in Adipozyten, Fibroblasten und Endothelzellen vorhanden, und Caveolin-3 ist nur im Skelett- und Herzmuskel vorhanden13, 15. Caveolin-1 ist das wichtigste und am meisten untersuchte. Es wird in zwei verschiedenen Isoformen (1a und 1b) ausgedrückt. Caveolin-1 transloziert von der Plasmamembran zum Lipidtröpfchen und ist für den Lipidhandel und den Stoffwechsel notwendig16. Lipidtröpfchen speichern Triglyceride nach der Fütterung und diese Moleküle werden zu Fettsäure hydrolysiert und während des Fastens freigesetzt; Dieser Mechanismus kann durch Caveolin-116 reguliert werden. Caveolin-1-Mangel erhöht auch die Anfälligkeit für Zelltod durch Autophagie17.
Das Gen CAVIN1 kodiert für ein zytoplasmatisches Protein namens Caveolae associated protein 1 (Cavin-1)14, 16, das für die Bildung und Stabilisierung von Caveolen obligatorisch ist. Cavin-1 wird in Adipozyten, Muskelzellen und anderen Zellen exprimiert und ist auch für die Übertragung von Caveolae-Signalen essentiell14, 18. Knockout des CAV1-Gens verursacht einen Mangel an Caveolae in Nicht-Muskelzellen, während der Knockout von CAVIN1 das Fehlen von Caveolae in allen Geweben, einschließlich muscle14, verursacht. Das Fehlen von Caveolen kann die Regulation der Lipolyse, des Fettsäureflusses, der Triglyceridsynthese und der Signale anderer Signalwege beeinflussen.
Arten von CGL
Basierend auf nachweisbaren genetischen Veränderungen werden vier Arten beschrieben. Die Typen 1 und 2 sind für über 95% der Fälle verantwortlich, und Typ 2 hat einen stärker betroffenen Phänotyp. Nur ein Fall von Typ 3 und rund 30 Fälle von Typ 4 wurden gemeldet4.
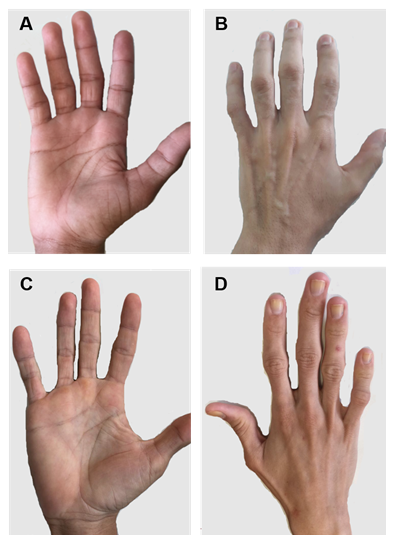
Abbildung 2. Hände von Patienten mit CGL Typ 1 und 2. (A) und (B) Anteriore und posteriore Ansichten der Hände von Typ-1-Patienten. Anscheinend normale Hände, da noch mechanisches Fettgewebe vorhanden ist. (C) und (D) Anteriore und posteriore Ansichten der Hände von Typ-2-Patienten. Die Schwere der Erkrankung ist größer, und der Mangel an Fett ist offensichtlich und leicht spürbar.
CGL Typ 1. Im Jahr 1999 haben Garg et al. beschrieben Patienten Mutation auf Chromosom 9q34, und drei Jahre später Agarwal et al. zeigte AGPAT2 als das von dieser Mutation2 betroffene Enzym, 19. Aufgrund der Mutation dieses AGPAT2 erfolgt keine oder nur eine minimale Produktion von Triacylglycerin durch den Stimulus anderer Isoformen. Der Phänotyp von AGPAT2-Knockout-Mäusen ähnelt dem von Menschen mit CGL-Typ, was die Rolle dieses Enzyms in der Pathophysiologie bestätigt20, 21.
CGL Typ 2. Magre et al. waren die ersten, die die Mutation im Seipin-Gen (Chromosom 11q13) identifizierten3. Mutationen (meist Unsinn) des Seipin-Gens (BSCL2) erzeugen ein verkürztes Protein und können den Lipidstoffwechsel durch verschiedene Mechanismen beeinflussen: a) Abnahme der Seipin-Stabilität; b) Verringerung der Fähigkeit, Lipin 1 zu binden; und c) Versagen, sich ausschließlich an die ER-Membran zu oligomerisieren und zu lokalisieren11. Einige Zellen sind immer noch in der Lage, Triacylglycerin und kleine Lipidtröpfchen zu erzeugen, aber große Lipidtröpfchen fehlen aufgrund des Verlustes der Fusionsfähigkeit dieser kleinen Lipidtröpfchen. Es gibt auch ein Versagen bei der Expression adipogener Faktoren wie dem Peroxisom-Proliferator-aktivierten Rezeptor-Gamma (PPARG) sowie Adiponektin und Adipozyten-Fettsäure-Bindungsprotein (FABP4)11, 16. Seipinmangel beeinträchtigt die Adipogenese, erhöht die Lipolyse und verhindert die Ansammlung von Triglyceriden in Adipozyten.
CGL Typ 3. Dieser Typ wurde kürzlich bei einem Patienten beschrieben, der trotz des CGL-Phänotyps keine Mutationen in den Genen AGPAT2 oder BSCL222 aufwies. Mäuse mit einer Mutation in Cav1 sind resistent gegen Diät-induzierte Fettleibigkeit und haben Insulinresistenz, Hypertriglyceridämie, vermindertes Adiponektin, reduzierte Fettmasse und kleine Adipozyten16. Nach der Auswahl von Kandidatengenen basierend auf Studien an Mäusen, Kim et al. bestätigte das Vorhandensein einer Nonsense-Mutation im Caveolin-1-Gen (CAV1) auf Chromosom 7q3122.
CGL Typ 4. In diesem seltenen Typ ist das betroffene Gen das CAVIN1, das für das Protein Cavin-1 kodiert. Beim Menschen wurde es bei Patienten mit generalisierter kongenitaler Lipodystrophie und Muskeldystrophie15, 23 berichtet.
Kürzlich wurden auch Mutationen in den Genen PCYT1A und PPARG beschrieben, die Lipodystrophye verursachen24, 25.
Klinische Merkmale
CGL-Patienten weisen normalerweise Akromegaloidfazies, Acanthosis nigricans, Phebomegalie, Hepatomegalie und Muskelhypertrophie5, 26, 27 auf. Mehrere Autoren zitieren Nabelbruch als klinischen Befund des Syndroms26. Wir haben die Häufigkeit bei unseren Patienten ausgewertet, und keiner von ihnen hat diese Änderung vorgelegt28. Tatsächlich verursacht das Fehlen von periumbilikalem Fettgewebe eine Protrusion der Nabelnarbe, und dies kann fälschlicherweise als Hernie diagnostiziert werden28, 29.
Sobald Adipozyten Fett nicht ausreichend speichern können, reichert es sich in anderen Geweben wie Leber und Muskeln an und verursacht eine schwere Insulinresistenz. Die Knochendichtemessung (DXA) kann eine normale oder hohe Knochenmineraldichte zeigen30 und reduziertes Gesamtkörperfett (normalerweise unter 6%)27. Als Folge der niedrigen Körperfett, Serum Adiponektin und Leptin sind niedrig too27. Da Leptin für die Kontrolle des Hungers unerlässlich ist, haben diese Patienten typischerweise Hyperphagie, die seit ihrer Kindheit leicht erkennbar ist. Adiponektin spielt eine wichtige Rolle als Insulinsensibilisator, und sein Mangel verschlechtert die Insulinresistenz. Trotzdem sind Glukose und glykiertes Hämoglobin zunächst auf Kosten sehr hoher Insulinspiegel normal. Diabetes beginnt normalerweise in der Pubertät; In unserer Serie betrug das mittlere Erkrankungsalter 15,8 ± 7,1 Jahre27. Anfangs werden sie mit oralen Medikamenten kontrolliert, die in einigen Jahren hohe Insulindosen benötigen27. Arterielle Hypertonie tritt bei einem Drittel der Patienten auf27.
Es gibt einige spezifische klinische Merkmale jedes CGL-Typs. Patienten mit Typ 1 weisen immer noch mechanisches Fettgewebe auf, insbesondere in Handflächen, Fußsohlen, orbitalen und periartikulären Bereichen31. Im Gegensatz dazu zeigen Typ-2-Patienten ein Fehlen von metabolischem und mechanischem Fettgewebe. Seipin wird im Gehirn und Kleinhirn stark exprimiert und ist auch an der Regulation neuronaler Funktionen beteiligt. Mehr als die Hälfte der Typ-2-Patienten hat eine kognitive Beeinträchtigung1, 8. Typen 3 und 4 haben die Erhaltung von mechanischem und Knochenmarkfett, und Typ 4 hat Muskelschwäche im Zusammenhang mit hoher Serum-Kreatinkinase und spinaler Instabilität15.
Es gibt auch geschlechtsspezifische klinische Merkmale. Polyzystische Ovarien und Amenorrhoe sind häufig32. Menstruationszyklen normalisieren sich normalerweise unter Verwendung von Metreleptin, wahrscheinlich aufgrund einer Verbesserung der Insulinsensitivität und Wiederherstellung der LH-Pulsatilität32. Typ-2-Männer können aufgrund des Mangels an Seipin in Keimzellen Teratozoospermie haben33.
Hypertriglyceridämie tritt seit den ersten Lebensjahren auf und kann eine akute Pankreatitis verursachen. HDL ist normalerweise niedriger als 30 mg / dl. Erhöhungen der Leberenzyme sind ebenfalls ein früher Befund und stammen von der Fettablagerung in der Leber. Eine fortschreitende Senkung der Blutplättchen im Serum deutet auf eine Verschlechterung der Lebererkrankung und eine wahrscheinliche Zirrhose34 hin.
Da Cavin-1 in den Muskelzellen vorhanden ist, haben Patienten mit Typ 4 eine leichte Muskelschwäche und eine erhöhte Kreatinkinase15.
Die Lebenserwartung, hauptsächlich bei Typ 2, ist erheblich gesunken, wobei der Tod nicht selten vor dem Alter von 30 Jahren auftritt (persönliche Erfahrung basierend auf 20 Patienten, die in den letzten 19 Jahren gestorben sind). Die Todesursachen hängen mit Diabetes (Nierenversagen, plötzlicher Tod), Leber (Leberzirrhose, Verdauungsblutungen) oder Infektionen zusammen.
Diagnose und Behandlung
Die CGL-Diagnose basiert auf klinischen Daten: Akromegaloidmerkmale, Acanthosis nigricans, Reduktion des gesamten Körperfetts, Muskelhypertrophie und Protrusion der Nabelschnurnarbe. Labordaten können auch Diabetes mit schwerer Insulinresistenz und Hypertriglyceridämie zeigen. Bildgebende Untersuchungen können helfen, ektopische Fettablagerungen hauptsächlich in Leber und Bauchspeicheldrüse (Lebersteatose mit Hepatomegalie und Pankreassteatose) zu identifizieren. Die DXA kann das niedrige Körperfett und die hohe Knochendichte30 bestätigen.
Die Behandlung von CGL besteht aus einer strengen Kontrolle der Ernährung mit einer Verringerung der Aufnahme von Fett, Kohlenhydraten, Triglyceriden und Lebensmitteln mit einem hohen glykämischen Index zur Vorbeugung und Kontrolle von Begleiterkrankungen29. Die ideale Ernährung ist jedoch aufgrund des gesteigerten Appetits und der befürworteten starken Einschränkung ein herausforderndes Ziel. Körperliche Aktivität sollte auch gefördert werden, um die Kontrolle von Komorbiditäten zu verbessern, außer bei Patienten mit Kontraindikationen wie schwerer Kardiomyopathie29.
In Bezug auf die medikamentöse Behandlung können diese Patienten mit den üblichen Medikamenten gegen Diabetes, Bluthochdruck und Dyslipidämie behandelt werden. Die erste Wahl für die Behandlung von Diabetes und Insulinresistenz ist Metformin, aber in der Regel ist es nicht genug. Im Gegensatz zur Behandlung der partiellen Lipodystrophie sollten Thiazolidindione mit Vorsicht angewendet werden29. Andere orale Antidiabetika werden verwendet, aber sie wurden nicht speziell bei CGL-Patienten untersucht. Es liegen Daten bei Tieren vor, die darauf hindeuten, dass die Anwendung von SGLT2-Inhibitoren (Dapagliflozin) Vorteile bei der Vorbeugung von Kardiomyopathien haben kann35; Studien sind erforderlich, um dies beim Menschen zu bestätigen. Wenn die Krankheit fortschreitet und eine schwere Insulinresistenz auftritt, sind hohe tägliche Insulindosen erforderlich. Das Fehlen von subkutanem Fettgewebe ist ein Problem bei der Verabreichung der hohen Insulindosen. Möglicherweise ist konzentrierteres Insulin (U-300 oder U500) erforderlich36. Diese Patienten weisen eine schwere Dyslipidämie auf, hauptsächlich aufgrund des Anstiegs von Triglyceriden und niedrigem HDL, und daher ist manchmal die Verwendung von Fibrat erforderlich, um eine akute Pankreatitis zu verhindern. Darüber hinaus sollte aufgrund des hohen kardiovaskulären Risikos dieser Patienten eine Intervention mit einem Statin in Betracht gezogen werden, und die Ziele von LDL oder Nicht-HDL sollten streng sein29.
Tägliche Injektionen von Metreleptin verursachen eine signifikante Abnahme des Appetits und bringen Vorteile durch Senkung von Glykämie, Triglyceridämie und Leberenzymen. Besonders bei Kindern ist die Verringerung des Bauchumfangs bemerkenswert, wahrscheinlich aufgrund einer Verringerung der Hepatomegalie.
Schlussfolgerung
CGL ist eine seltene und schwere Erkrankung, die bei Diabetes (normalerweise mit hohen Insulindosen) und frühem Tod auftreten kann. Der Phänotyp des Patienten ist ziemlich charakteristisch und erfordert jedoch die Kenntnis des Syndroms durch die Angehörigen der Gesundheitsberufe, um eine frühzeitige Diagnose zu stellen. Metreleptin scheint derzeit das einzige Medikament zu sein, das den natürlichen Krankheitsverlauf verändern kann.
Interessenkonflikt: keine.
- N. T. Erforschung der Pathophysiologie hinter den häufigeren genetischen und erworbenen Lipodystrophien. Zeitschrift für Humangenetik. 2014 Januar; 59(1): 16-23.
- Agarwal AK, Arioglu E, De Almeida S, et al. AGPAT2 ist bei angeborener generalisierter Lipodystrophie mutiert, die mit Chromosom 9q34 verbunden ist. Nat Genet. 2002 Mai; 31(1): 21-3.
- Magre J, Delepine M, Khallouf E, et al. Identifizierung des Gens verändert in Berardinelli-Seip angeborene Lipodystrophie auf Chromosom 11q13. Naturgenetik. 2001 August; 28(4): 365-70.
- Patni N, Garg A. Angeborene generalisierte Lipodystrophien – neue Einblicke in metabolische Dysfunktion. Nature reviews Endokrinologie. 2015 September; 11(9): 522-34.
- Garg A. Erworbene und vererbte Lipodystrophien. Das New England Journal of Medicine. 2004 Beschädigen 18; 350(12): 1220-34.
- de Azevedo Medeiros LB, Candido Dantas VK, Craveiro Sarmento AS, et al. Hohe Prävalenz der angeborenen Berardinelli-Seip-Lipodystrophie im Bundesstaat Rio Grande do Norte im Nordosten Brasiliens. In: Diabetol Metab Syndr. 2017; 9: 80.
- Chiquette E, Oral EA, Garg A, et al. Schätzung der Prävalenz von generalisierter und partieller Lipodystrophie: Erkenntnisse und Herausforderungen. Diabetes, metabolisches Syndrom und Fettleibigkeit: Ziele und Therapie. 2017: 375-83.
- Wee K, Yang W, Sugii S, et al. Auf dem Weg zu einem mechanistischen Verständnis von Lipodystrophie und Seipin-Funktionen. In: Bioscience reports. 2014; 34(5).
- Dollet L, Magre J, Cariou B, et al. Funktion von Seipin: Neue Erkenntnisse aus Bscl2/Seipin Knockout-Mausmodellen. Biochimie. 2014 Januar; 96: 166-72.
- Sim MF, Dennis RJ, Aubry EM, et al. Das humane Lipodystrophieprotein Seipin ist ein ER-Membranadapter für die adipogene PA-Phosphatase Lipin1. Molekularer Stoffwechsel. 2012; 2(1): 38-46.
- Sim MF, Talukder MM, Dennis RJ, et al. Die Analyse von natürlich vorkommenden Mutationen im humanen Lipodystrophie-Protein Seipin zeigt mehrere potenzielle pathogene Mechanismen. In: Diabetologia. 2013 November; 56(11): 2498-506.
- Payne VA, Grimsey N, Tuthill A, et al. Das humane Lipodystrophie-Gen BSCL2/seipin kann für eine normale Adipozytendifferenzierung essentiell sein. Diabetes. 2008 August; 57(8): 2055-60.
- Cohen AW, Hnasko R, Schubert W, et al. Rolle von Caveolae und Caveolinen in Gesundheit und Krankheit. Physiologische Bewertungen. 2004 Oktober; 84(4): 1341-79.
- Pilch PF, Liu L. Fetthöhlen: Hohlräume, Lipidhandel und Lipidstoffwechsel in Adipozyten. Trends in Endokrinologie und Stoffwechsel: TEM. 2011 August; 22(8): 318-24.
- Hayashi YK, Matsuda C, Ogawa M, et al. Menschliche PTRF-Mutationen verursachen einen sekundären Mangel an Caveolinen, was zu einer Muskeldystrophie mit generalisierter Lipodystrophie führt. In: J Clin Invest. 2009 September; 119(9): 2623-33.
- Parton RG, del Pozo MA. Caveolae als Plasmamembransensoren, Protektoren und Organisatoren. Nature reviews Molekulare Zellbiologie. 2013 Februar; 14(2): 98-112.
- Le Lay S, Briand N, Blouin CM, et al. Das lipoatrophische Caveolin-1-defiziente Mausmodell zeigt Autophagie in reifen Adipozyten. Autophagie. 2010 August; 6(6): 754-63.
- Liu L, Brown D, McKee M, et al. Die Deletion von Cavin / PTRF verursacht einen globalen Verlust von Caveolen, Dyslipidämie und Glukoseintoleranz. Zellstoffwechsel. 2008 Oktober; 8(4): 310-7.
- Garg A, Wilson R, Barnes R, et al. Ein Gen für angeborene generalisierte Lipodystrophie ist dem menschlichen Chromosom 9q34 zugeordnet. Das Journal für klinische Endokrinologie und Stoffwechsel. 1999 September; 84(9): 3390-4.
- Vogel P, Lesen R, Hansen G, et al. Pathologie der kongenitalen generalisierten Lipodystrophie bei Agpat2-/- Mäusen. Veterinärmedizinische Pathologie. 2011 Mai; 48(3): 642-54.
- Cortes VA, Curtis DE, Sukumaran S, et al. Molekulare Mechanismen der Lebersteatose und Insulinresistenz im AGPAT2-defizienten Mausmodell der kongenitalen generalisierten Lipodystrophie. Zellstoffwechsel. 2009 Februar; 9(2): 165-76.
- Kim CA, Delepine M, Boutet E, et al. Assoziation einer homozygoten Nonsense-Caveolin-1-Mutation mit kongenitaler Berardinelli-Seip-Lipodystrophie. In: J Clin Endocrinol Metab. 2008 April; 93(4): 1129-34.
- Rajab A, Straub V, McCann LJ, et al. Tödliche Herzrhythmusstörungen und Long-QT-Syndrom in einer neuen Form der angeborenen generalisierten Lipodystrophie mit Muskelrippeln (CGL4) aufgrund von PTRF-CAVIN-Mutationen. PLoS genetics. 2010 Beschädigen 12; 6(3): e1000874.
- Payne F, Lim K, Girousse A, et al. Mutationen, die den metabolischen Phosphatidylcholinweg bei Menschen mit angeborener Lipodystrophie und Fettlebererkrankung stören. Proc Natl Acad Sci USA 2014 Juni 17; 111(24): 8901-6.
- Dyment DA, Gibson WT, Huang L, et al. Biallelische Mutationen bei PPARG verursachen eine angeborene, generalisierte Lipodystrophie ähnlich dem Berardinelli-Seip-Syndrom. In: Eur J Med Genet. 2014 September; 57(9): 524-6.
- Garg A. Clinical review#: Lipodystrophien: genetische und erworbene Körperfettstörungen. Das Journal für klinische Endokrinologie und Stoffwechsel. 2011 November; 96(11): 3313-25.
- Lima JG, Nobrega LH, de Lima NN, et al. Klinische und Labordaten einer großen Reihe von Patienten mit angeborener generalisierter Lipodystrophie. In: Diabetol Metab Syndr. 2016; 8: 23.
- Lima GJ, Lima NN, Oliveira CF, et al. Nabelbruch bei Patienten mit Berardinelliseip-Syndrom: Ist es wirklich Hernie. In: J Clin Mol Endocrinol. 2015; 1(1): 3.
- Braun RJ, Araujo-Vilar D, Cheung PT, et al. Die Diagnose und Behandlung von Lipodystrophie-Syndromen: Eine multigesellschaftliche Praxisrichtlinie. In: J Clin Endocrinol Metab. 2016 Dezember; 101(12): 4500-11.
- Lima JG, Nobrega LH, Lima NN, et al. Die Knochendichte bei Patienten mit angeborener Berardinelli-Seip-Lipodystrophie ist an trabekulären Stellen und bei Typ-2-Patienten höher. J Clin Densitom. 2016 November 25.
- Simha V, Garg A. Phänotypische Heterogenität der Körperfettverteilung bei Patienten mit angeborener generalisierter Lipodystrophie, verursacht durch Mutationen in den AGPAT2- oder Seipin-Genen. In: J Clin Endocrinol Metab. 2003 November; 88(11): 5433-7.
- Musso C, Cochran E, Javor E, et al. Die Langzeitwirkung der rekombinanten Methionyl-Human-Leptin-Therapie auf Hyperandrogenismus und Menstruationsfunktion bei Frauen und Hypophysenfunktion bei männlichen und weiblichen hypoleptinämischen lipodystrophischen Patienten. Stoffwechsel. 2005 Februar; 54(2): 255-63.
- Jiang M, Gao M, Wu C, et al. Der Mangel an testikulärem Seipin verursacht bei Männern ein Teratozoospermie-Syndrom. Proc Natl Acad Sci USA 2014 Mai 13; 111(19): 7054-9.
- Mitchell O, Feldman DM, Diakow M, et al. Die Pathophysiologie der Thrombozytopenie bei chronischen Lebererkrankungen. In: Hepat Med. 2016; 8: 39-50.
- Joubert M, Jagu B, Montaigne D, et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes. 2017 Apr; 66(4): 1030-40.
- Lima JG, Lima NN, Lima RLM, et al. Glargine U300 Insulin as a Better Option than Degludec U100 to Treat a Congenital Generalized Lipodystrophy Patient. Clin Diabetes Res. 2017; 1(1).