Článek
Josivan Gomes Lima1*, Marcel Catão Ferreira dos Santos1, Julliane Tamara de Araújo Melo Campos2
1Departamento de medicina kliniky, disciplina de endocrinologia e metabologia. Hospital Universitário Onofre Lopes, Universidade Federal do Rio Grande do Norte (UFRN), Natal, RN, Brazílie
2Faculty Zdravotnických Věd Trairi, Federal University of Rio Grande do Severní (UFRN), Natal, RN, Brazílie
Abstrakt
Vrozená Generalizovaná Lipodystrofie (CGL) je vzácné a závažné autozomálně recesivní onemocnění. Pacienti jsou vadní při ukládání tělesného tuku a v důsledku toho ukládají tuk do ektopických tkání, zejména jater,a mohou vyvinout cirhózu. Inzulínová rezistence je typickým nálezem, který způsobuje cukrovku, která vyžaduje vysoké denní dávky inzulínu. Ve státě Rio Grande do Norte v Brazílii máme jednu z největších kohort pacientů s CGL. V tomto článku přezkoumáváme patofyziologii, klinický obraz a léčbu této nemoci.
Úvod
diabetes Typu 2 je světový zdravotní problém, a obvykle výsledky z nadměrné hmotnosti a zvýšení viscerálního tuku způsobuje periferní inzulínovou rezistenci a neschopnost slinivky břišní k uvolňování inzulínu, aby kompenzovat tuto rezistenci. Další méně časté typy diabetu dojít v důsledku specifických genetických mutací, jako Vrozená Generalizovaná Lipodystrofie (CGL), také známý jako Berardinelli-Seip Vrozené Lipodystrofie (BSCL). CGL je autozomálně recesivní onemocnění, které je rozděleno do čtyř typů na základě genové mutace. Změněné geny hrají základní funkce pro tvorbu adipocytů, produkci lipidů a správné ukládání uvnitř adipocytů. Mutace snížit tukové tkáně s následným ukládáním tuku v mimoděložní míst, což způsobuje tuk, játra, změněný metabolismus sacharidů, závažná inzulínová rezistence s hyperinzulinémií a acromegaloid funkce, a dyslipidemia1-3. Syndrom CGL má na světě hlášeno kolem 500 případů. V Brazílii, ve státě Rio Grande do Norte (RN), jsme diagnostikovali, léčili a sledovali 54 případů za posledních 20 let4, 5. V popisné studii s využitím sekundárních dat jsme odhadli celkem 103 pacientů v RN6. To naznačuje mnohem vyšší prevalenci než prevalence uvedená v literatuře (1: 1 mil.) 7.
Triacylglycerolu tvorbě a ukládání v tukové kapénky
biosyntézy triglyceridů a fosfolipidů (Obrázek 1A) začíná s glycerol-3-fosfát-acyltransferase (GPAT) acylating glycerol-3-fosfátu v pozici 1, tvoří 1-Acylglycerol-3-fosfát (lysophosphatidic kyseliny). Následuje další acylace krok na pozici dva enzymem AGPAT (1-Acylglycerol-3-fosfát-acyltransferase), pocházející 1,2-Diacylglycerolový-3-fosfát (fosfatidová kyselina). Je to klíčový mezistupeň v cestě biosyntézy triglyceridů i fosfoglyceridů. Existuje 11 izoform agpat enzymů, kódovaných různými geny4. AGPAT1 a AGPAT2 jsou nejrozsáhleji studovány. AGPAT1 je přítomen ve vysokých hladinách varlat, slinivky břišní a v menší míře v tukové tkáni a dalších tkáních, jako je srdce, placenta, mozek, plíce, zatímco AGPAT2 je hojný v tukové tkáni. V následujících krocích, cytosolový enzym fosfatidových kyselé fosfatázy (PAP nebo lipin) pochází 1,2-diacylglycerolový, a 1,2-diacylglycerolový acyltransferase (DGAT) formy triacylglycerol4. Kyselina fosfatidová a diacylglycerol mohou také pocházet z jiných fosfolipidů, jako je kardiolipin, fosfatidylinositol a fosfatidylcholin.
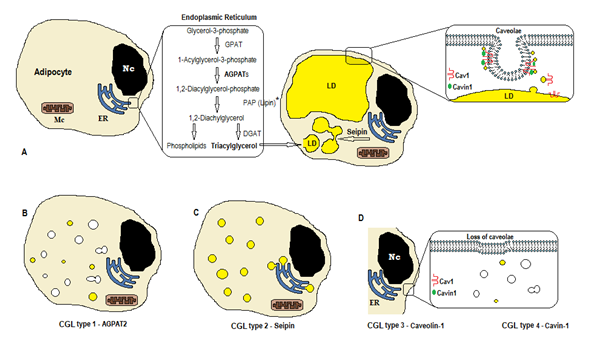
Obrázek 1. Schéma syntézy triglyceridů podle typů CGL. A) normální syntéza a skladování triacylglycerolu (TAG) v adipocytech. (B) mutace AGPAT2 snižuje produkci tagů (některé jsou stále syntetizovány pod stimulací jiných Agpatů). (C) mutace seipinového genu snižuje syntézu tagů a tvorbu a fúzi lipidových kapiček (LD). (D) Caveolin-1 a Cavin-1 jsou potřebné pro tvorbu a stabilizaci caveolae. Mutace v CAV1 (typ 3) nebo CAVIN1 (typ 4), může způsobit ztrátu caveolae v membráně. Nc, nucleus. Endoplazmatické retikulum. MC, mitochondrie. * Lipin je cytosolický enzym ukotvený seipinem v ER.
tyto reakce se vyskytují v endoplazmatickém retikulu adipocytů (ER), kde progresivní akumulace triglyceridů způsobuje tvorbu malých lipidových kapiček (LD)8. Produkt genu BSCL2 je transmembránový protein zvaný seipin, který způsobuje fúzi malého LD, pocházejícího z velkého LD. Seipin sídlí v ER a soustředí se na křižovatce s rodící LD, usnadnění lipidů provoz mezi ER a LD a začlenění triglyceridů v LD9. Seipin může také působit jako ER kotva k cytosolickému enzymu lipin 1. Kromě toho, že je nezbytné pro lipidové kapičky fusion, velikosti a morfologie, seipin je také nezbytný pro adipogenesis (prostřednictvím interakce lipin 1) a mobilní triglyceridů lipolysis10, 11. Nedostatek seipinu brání diferenciaci preadipocytů na adipocyty a ovlivňuje konečné zrání9, jak ukazují studie na mezenchymálních kmenových buňkách s BSCL2 vyřazeny12. Nemastné tkáně také exprimují seipin a je třeba určit další funkce.
V adipocytech, caveolae, které jsou specializované 50-100nm membránových invaginací, tvoří 20% z plazmatické membrány oblasti, tvorba adipocytů, buněk s nejvyšší hustotou caveolae13. Tvorba lipidových kapiček potřebuje membránový protein (Caveolin-hlavní složka membrán caveolae) a cytoplazmatický protein (Cavin-1)14. Geny CAV1, CAV2 a CAV3 kódují tři formy caveolinu s podobnými strukturami (Caveolin-1, Caveolin-2 a Caveolin-3). Caveolin-1 a Caveolin-2 jsou přítomny v adipocytech, fibroblastů a endoteliálních buněk, a Caveolin-3 je přítomen pouze v kosterním a srdečním muscle13, 15. Caveolin-1 je nejdůležitější a nejvíce studovaný. Vyjadřuje se ve dvou různých izoformách (1a a 1b). Caveolin-1 se translokuje z plazmatické membrány na lipidové kapičky, což je nezbytné pro obchodování s lipidy a metabolismu16. Lipidové kapičky ukládání triglyceridů po krmení a tyto molekuly jsou hydrolyzovány na mastné kyseliny a uvolňuje během půstu; tento mechanismus může být regulována Caveolin-116. Nedostatek Caveolin-1 také zvyšuje náchylnost k buněčné smrti autofagií17.
gen CAVIN1 kóduje cytoplazmatický protein zvaný caveolae associated protein 1 (Cavin-1)14, 16, které je povinné pro tvorbu a stabilizaci caveolae. Cavin-1 je exprimován v adipocytech, svalové buňky a další buňky, a je také nezbytný při přenosu caveolae-vznikl signals14, 18. Knockout CAV1 genu způsobuje nedostatek caveolae v non-svalové buňky, vzhledem k tomu, že knockout CAVIN1 příčiny absence caveolae ve všech tkáních, včetně muscle14. Nedostatek caveolae může ovlivnit regulaci lipolýzy, toku mastných kyselin, syntézu triglyceridů a signály jiných cest.
typy CGL
na základě detekovatelných genetických změn jsou popsány čtyři typy. Typy 1 a 2 jsou zodpovědné za více než 95% případů a typ 2 má závažněji postižený fenotyp. Byl hlášen pouze jeden případ typu 3 a přibližně 30 případů typu 44.
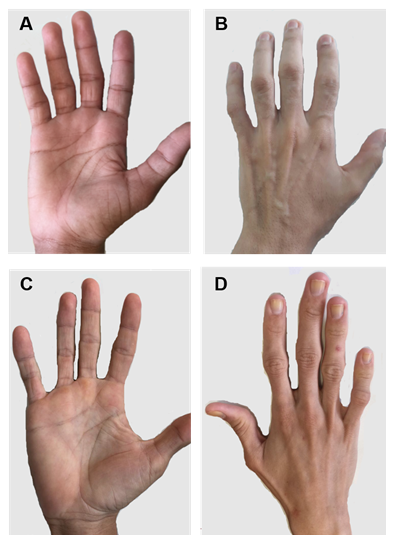
Obrázek 2. Ruce pacientů s CGL typu 1 a 2. (A) A (B) přední a zadní pohled na ruce pacientů typu 1. Zřejmě normální ruce, protože stále existuje mechanická tuková tkáň. (C) A (D) přední a zadní pohled na ruce pacientů typu 2. Závažnost onemocnění je větší a nedostatek tuku je zřejmý a snadno patrný.
CGL typ 1. V roce 1999 Garg et al. popsaná mutace pacientů na chromozomu 9q34 a o tři roky později Agarwal et al. ukázal AGPAT2 jako enzym ovlivněný touto mutací2, 19. V důsledku mutace tohoto AGPAT2 nedochází k žádné nebo minimální produkci triacylglycerolu stimulací jiných izoform. Fenotyp knockoutových myší AGPAT2 je podobný fenotypu lidí s typem CGL, což potvrzuje roli tohoto enzymu v patofyziologii20, 21.
CGL typ 2. Magre a kol. byli první, kdo identifikoval mutaci v genu seipin (chromozom 11q13)3. Mutace (většinou nesmysl) seipin gen (BSCL2) produkují zkrácený protein a může mít vliv na metabolismus lipidů tím, že různými mechanismy: a) snížení seipin stabilitu; b) snížení schopnosti vázat lipin 1; a c) selhání oligomerize a lokalizovat sám výhradně na POHOTOVOST membrane11. Některé buňky jsou stále schopny generovat triacylglycerol a malé lipidové kapičky, ale velké lipidové kapičky chybí kvůli ztrátě schopnosti fúze těchto malých lipidových kapiček. Existuje také selhání exprese adipogenních faktorů, jako je receptor gama aktivovaný peroxizomovým proliferátorem (PPARG), stejně jako adiponektin a protein vázající mastné kyseliny adipocytů (FABP4)11, 16. Nedostatek seipinu zhoršuje adipogenezi, zvyšuje lipolýzu a zabraňuje akumulaci triglyceridů v adipocytech.
CGL typ 3. Tento typ byl nedávno popsán u pacienta, který navzdory fenotypu CGL neměl mutace v genech AGPAT2 nebo BSCL222. Myši s mutací v Cav1 jsou odolné vůči dietou indukované obezity a inzulínové rezistence, hypertriglyceridémie, snížení adiponektinu, snižuje množství tuku, a malé adipocytes16. Po výběru kandidátských genů na základě studií na myších, Kim et al. potvrdila přítomnost nesmyslné mutace v genu caveolin-1 (CAV1) na chromozomu 7q3122.
CGL typ 4. U tohoto vzácného typu je postiženým genem CAVIN1, který kóduje protein Cavin-1. U lidí to bylo hlášeno u pacientů s generalizovanou vrozenou lipodystrofií a svalovou dystrofií15, 23.
nedávno byly také popsány mutace v genech PCYT1A a PPARG způsobující lipodystrofii24, 25.
Klinické funkce
CGL pacienti obvykle přítomen acromegaloid facie, akantóza, phebomegaly, hepatomegalie, a svalové hypertrophy5, 26, 27. Několik autorů uvádí pupeční kýlu jako klinický nález syndromu26. Vyhodnocovali jsme jeho četnost u našich pacientů a nikdo z nich tuto změnu nepředložil. Ve skutečnosti nepřítomnost periumbilické tukové tkáně způsobuje výčnělek pupeční jizvy, což může být mylně diagnostikováno jako kýla28, 29.
jakmile adipocyty nemohou dostatečně ukládat tuk, hromadí se v jiných tkáních, jako jsou játra a svaly, což způsobuje silnou inzulínovou rezistenci. Kostní denzitometrie (DXA)může vykazovat normální nebo vysokou kostní minerální hustotu30 a snížený celkový tělesný tuk (obvykle nižší než 6%) 27. V důsledku nízkého tělesného tuku je také nízký adiponektin v séru a leptin27. Protože leptin je nezbytný při kontrole hladu, tito pacienti mají obvykle hyperfágii, což je zřejmé již od dětství. Adiponektin hraje důležitou roli jako senzibilizátor inzulínu a jeho nedostatek zhoršuje inzulínovou rezistenci. Navzdory tomu jsou zpočátku glukóza a glykovaný hemoglobin normální na úkor velmi vysokých hladin inzulínu. Diabetes obvykle začíná v pubertě; v naší sérii byl průměrný věk nástupu 15,8±7,1 let27. Zpočátku jsou kontrolovány perorálními léky, které potřebují vysoké dávky inzulinu za několik let27. Arteriální hypertenze se vyskytuje u jedné třetiny pacientů27.
existují určité specifické klinické příznaky každého typu CGL. Pacienti s typem 1 stále přítomni mechanický tukový tuk, zejména v dlaních, chodidlech, orbitálních, periartikulárních oblastech31. Naproti tomu pacienti typu 2 vykazují nepřítomnost metabolických a mechanických tukových tkání. Seipin je vysoce exprimován v mozku a cerebellum a podílí se také na regulaci nervových funkcí. Více než polovina pacientů typu 2 má určité kognitivní poruchy1, 8. Typy 3 a 4 je zachování mechanické a kostní dřeně, tuku a typ 4 se svalová slabost, spojené s vysokým sérové kreatin kinázy a páteře instability15.
existují také genderově specifické klinické rysy. Polycystické vaječníky a amenorea jsou běžné32. Menstruační cykly se obvykle vrátí k normálu s použitím metreleptinu, pravděpodobně kvůli zlepšení citlivosti na inzulín a obnovení pulsatility32. Muži typu 2 mohou mít teratozoospermii kvůli nedostatku seipinu v zárodečných buňkách33.
hypertriglyceridemie se vyskytuje od prvních let života a může způsobit akutní pankreatitidu. HDL je obvykle nižší než 30 mg / dl. Zvýšení jaterních enzymů je také časným nálezem a pochází z ukládání tuku v játrech. Progresivní snížení sérových krevních destiček naznačuje zhoršení jaterního onemocnění a pravděpodobnou cirhózu34.
protože Cavin-1 je přítomen ve svalových buňkách, pacienti s typem 4 mají mírnou svalovou slabost a zvýšenou kreatinkinázu15.
průměrná délka Života, především v typu 2, je podstatně snížil, se smrtí nezřídka dochází před dosažením věku 30 let (osobní zkušenost na základě 20 pacientů, kteří zemřeli v posledních 19 let). Příčiny úmrtí souvisejí s diabetem (selhání ledvin, náhlá smrt), játry (cirhóza, zažívací krvácení) nebo infekcemi.
Diagnostika a Léčba
CGL diagnóza je založena na klinických údajích: acromegaloid funkce, akantóza, snížení celkového tělesného tuku, svalové hypertrofie, a výstupek pupeční jizvu. Laboratorní údaje mohou také ukázat diabetes s těžkou inzulínovou rezistencí a hypertriglyceridemií. Zobrazovací testy mohou pomoci identifikovat ektopické usazeniny tuku hlavně v játrech a slinivce břišní (jaterní steatóza s hepatomegalií a pankreatickou steatózou). DXA může potvrdit nízký tělesný tuk a vysokou hustotu kostí30.
léčba CGL spočívá v přísné kontrole stravy s poklesem příjmu tuku, hlavně triglyceridů a potravin s vysokým glykemickým indexem k prevenci a kontrole komorbidit29. Nicméně, ideální dieta je náročné cíl dosáhnout z důvodu zvýšené chuti k jídlu a těžkou omezení obhajoval. Rovněž by měla být podporována fyzická aktivita, aby se zlepšila kontrola komorbidit, s výjimkou pacientů s kontraindikacemi, jako je těžká kardiomyopatie29.
pokud jde o léčbu drogami, mohou být tito pacienti léčeni obvyklými léky na diabetes, hypertenzi a dyslipidemii. První volbou pro léčbu diabetu a inzulínové rezistence je metformin, ale obvykle to nestačí. Na rozdíl od léčby částečné lipodystrofie by měly být thiazolidindiony používány s opatrností29. Používají se jiná perorální antidiabetika, ale nebyla specificky studována u pacientů s CGL. Tam jsou údaje u zvířat naznačují, že použití inhibitory SGLT2 (dapagliflozin) by mohl mít výhody prevenci cardiomyopathy35; studie jsou nezbytné pro potvrzení to v lidech. Jak onemocnění postupuje a dochází k závažné inzulínové rezistenci, jsou zapotřebí vysoké denní dávky inzulínu. Nedostatek podkožní tukové tkáně je problémem při podávání vysokých dávek inzulínu. Může být vyžadován koncentrovanější inzulín (u-300 nebo U500) 36. Tito pacienti představují závažné dyslipidémie, a to především v důsledku zvýšení triglyceridů a nízkými hladinami HDL, a proto, použití colestipol je někdy nutné, aby se zabránilo akutní pankreatitidy. Navíc vzhledem k vysokému kardiovaskulárnímu riziku u těchto pacientů by měla být zvážena intervence statinem a cíle LDL nebo non-HDL by měly být striktní29.
denní injekce metreleptinu způsobují významné snížení chuti k jídlu a přinášejí výhody snížením glykémie, triglyceridémie a jaterních enzymů. Zvláště u dětí je pozoruhodné snížení obvodu břicha, pravděpodobně v důsledku snížení hepatomegalie.
závěr
CGL je vzácné a závažné onemocnění, které se může objevit u diabetu (obvykle vyžadujícího vysoké dávky inzulínu) a předčasné smrti. Fenotyp pacienta je zcela charakteristický a vyžaduje však znalost syndromu zdravotnickými pracovníky k včasné diagnostice. Metreleptin se v současné době jeví jako jediný lék, který může změnit přirozenou historii onemocnění.
střet zájmů: Žádný.
- Nolis T. zkoumání patofyziologie za běžnějšími genetickými a získanými lipodystrofiemi. Žurnál lidské genetiky. 2014 leden; 59 (1): 16-23.
- Agarwal AK, Arioglu E, De Almeida S, et al. AGPAT2 je mutován v vrozené generalizované lipodystrofii spojené s chromozomem 9q34. Nat Genet. 2002 Květen; 31 (1): 21-3.
- Magre J, Delepine M, Khallouf E, et al. Identifikace genu pozměněného vrozenou lipodystrofií Berardinelli-Seip na chromozomu 11q13. Přírodní genetika. 2001 Srpen; 28 (4): 365-70.
- Patni N, Garg A. Vrozené generalizované lipodystrofie-nové poznatky o metabolické dysfunkci. Příroda recenze Endokrinologie. 2015 Září; 11 (9): 522-34.
- Garg a. získal a zdědil lipodystrofie. New England journal of medicine. 2004 Mar 18; 350 (12): 1220-34.
- de Azevedo Medeiros LB, Candido Dantas VK, Craveiro Sarmento AS, et al. Vysoká prevalence vrozené lipodystrofie Berardinelli-Seip ve státě Rio Grande do Norte, severovýchodní Brazílie. Diabetol Metab Syndr. 2017; 9: 80.
- Chiquette E, Oral EA, Garg A, et al. Odhad prevalence generalizované a částečné lipodystrofie: zjištění a výzvy. Diabetes, metabolický syndrom a obezita: cíle a terapie. 2017: 375-83.
- Wee K, Yang W, Sugii S, et al. Směrem k mechanickému porozumění lipodystrofii a seipinovým funkcím. Bioscience zprávy. 2014; 34(5).
- Dollet L, Magre J, Cariou B, et al. Funkce seipin: nové poznatky z modelů myši Bscl2/seipin knockout. Biochimie. 2014 leden; 96: 166-72.
- Sim MF, Dennis RJ, Aubry EM, et al. Lidský lipodystrofický protein seipin je membránový adaptér ER pro adipogenní pa fosfatázu lipin 1. Molekulární metabolismus. 2012; 2(1): 38-46.
- Sim MF, Talukder MM, Dennis RJ, et al. Analýza přirozeně se vyskytujících mutací v lidském lipodystrofickém proteinu seipin odhaluje několik potenciálních patogenních mechanismů. Diabetologie. 2013 Listopad; 56 (11): 2498-506.
- Payne va, Grimsey N, Tuthill a, et al. Gen lidské lipodystrofie BSCL2/seipin může být nezbytný pro normální diferenciaci adipocytů. Diabetes. 2008 srpnu; 57 (8): 2055-60.
- Cohen AW, Hnsko R, Schubert W, et al. Role caveolae a caveolins ve zdraví a nemoci. Fyziologické recenze. 2004 Říjen; 84 (4): 1341-79.
- Pilch PF, Liu L. tukové jeskyně: caveolae, obchodování s lipidy a metabolismus lipidů v adipocytech. Trendy v endokrinologii a metabolismu: TEM. 2011 srpnu; 22 (8): 318-24.
- Hayashi YK, Matsuda C, Ogawa M, et al. Lidské mutace PTRF způsobují sekundární nedostatek kaveolinů, což vede ke svalové dystrofii se generalizovanou lipodystrofií. J Clin Invest. 2009 září; 119 (9): 2623-33.
- Parton RG, del Pozo MA. Caveolae jako plazmatické membránové senzory, chrániče a organizéry. Příroda recenze molekulární buněčné biologie. 2013 Února; 14 (2): 98-112.
- Le Lay S, Briand N, Blouin CM, et al. Model myši s nedostatkem lipoatrofního caveolinu-1 odhaluje autofagii ve zralých adipocytech. Autofagie. 2010 Srpen; 6 (6): 754-63.
- Liu L, Brown D, McKee M, et al. Delece Cavin / PTRF způsobuje globální ztrátu caveolae, dyslipidemii a glukózovou intoleranci. Buněčný metabolismus. 2008 Říjen; 8 (4): 310-7.
- Garg A, Wilson R, Barnes R, et al. Gen pro vrozenou generalizovanou lipodystrofii mapuje lidský chromozom 9q34. Journal of clinical endocrinology and metabolism. 1999 září; 84 (9): 3390-4.
- Vogel P, Read R, Hansen G, et al. Patologie vrozené generalizované lipodystrofie u myší Agpat2 -/ -. Veterinární patologie. 2011 Smět; 48 (3): 642-54.
- Cortes va, Curtis DE, Sukumaran S, et al. Molekulární mechanismy jaterní steatózy a inzulínové rezistence v myším modelu s deficitem AGPAT2 vrozené generalizované lipodystrofie. Buněčný metabolismus. 2009 února; 9 (2): 165-76.
- Kim CA, Delepine M, Boutet E, et al. Asociace homozygotní nesmyslné mutace caveolin-1 s vrozenou lipodystrofií berardinelli-Seip. J Clin Endocrinol Metab. 2008 dubna; 93 (4): 1129-34.
- Rajab A, Straub V, McCann LJ, et al. Fatální srdeční arytmie a syndrom dlouhého QT v nové formě vrozené generalizované lipodystrofie se zvlněním svalů (CGL4) v důsledku mutací PTRF-CAVIN. PLoS genetics. 2010 Mar 12; 6 (3): e1000874.
- Payne F, Lim K, Girousse A, et al. Mutace narušující Kennedyho fosfatidylcholinovou dráhu u lidí s vrozenou lipodystrofií a tukovým onemocněním jater. Proc Natl Acad Sci U S A.2014 Června 17; 111(24): 8901-6.
- Dyment DA, Gibson WT, Huang L, et al. Bialelické mutace u PPARG způsobují vrozenou generalizovanou lipodystrofii podobnou syndromu Berardinelli-Seip. Eur J Med Genet. 2014 září; 57 (9): 524-6.
- Garg a. klinické hodnocení#: lipodystrofie: genetické a získané poruchy tělesného tuku. Journal of clinical endocrinology and metabolism. 2011 Listopad; 96 (11): 3313-25.
- Lima JG, Nobrega LH, de Lima NN, et al. Klinické a laboratorní údaje velké řady pacientů s vrozenou generalizovanou lipodystrofií. Diabetol Metab Syndr. 2016; 8: 23.
- Lima GJ, Lima NN, Oliveira CF, et al. Umbilická kýla u pacientů se syndromem Berardinelliseip: je to opravdu kýla. J Clin Mol Endokrinol. 2015; 1(1): 3.
- Brown RJ, Araujo-Vilar D, Cheung PT, et al. Diagnostika a léčba Lipodystrofických syndromů: směrnice pro více společností. J Clin Endocrinol Metab. 2016 Prosinec; 101 (12): 4500-11.
- Lima JG, Nobrega LH, Lima NN, et al. Hustota kostí u pacientů s vrozenou lipodystrofií Berardinelli-Seip je vyšší v trabekulárních místech a u pacientů typu 2. J Clin Densitom. 2016 Nov 25.
- Simha V, Garg a. fenotypová heterogenita v distribuci tělesného tuku u pacientů s vrozenou generalizovanou lipodystrofií způsobenou mutacemi v genech AGPAT2 nebo seipin. J Clin Endocrinol Metab. 2003 Listopad; 88 (11): 5433-7.
- Musso C, Cochran E, Javor E, et al. Dlouhodobý účinek rekombinantní methionyl, humánní růstový leptin terapie na hyperandrogenism a menstruačního funkce u žen a funkce hypofýzy v mužské a ženské hypoleptinemic lipodystrophic pacientů. Metabolismus. 2005 Únor; 54 (2): 255-63.
- Jiang M, Gao M, Wu C, et al. Nedostatek testikulárního seipinu způsobuje syndrom teratozoospermie u mužů. Proc Natl Acad Sci U S A.2014 13. Května; 111(19): 7054-9.
- Mitchell O, Feldman DM, Diakow M, et al. Patofyziologie trombocytopenie u chronického onemocnění jater. Hepat Med. 2016; 8: 39-50.
- Joubert M, Jagu B, Montaigne D, et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes. 2017 Apr; 66(4): 1030-40.
- Lima JG, Lima NN, Lima RLM, et al. Glargine U300 Insulin as a Better Option than Degludec U100 to Treat a Congenital Generalized Lipodystrophy Patient. Clin Diabetes Res. 2017; 1(1).