Autosomálně Recesivní Kongenitální Ichtyózy | Actas Dermo-Sifiliográficas
Úvod
nejnovější konsensus klasifikace ichtyóza rozlišuje 2 hlavní formy: nonsyndromic forem, které představují s kožní projevy pouze, a syndromový forem, které představují s projevy v jiných orgánech (Tabulka 1).1 mezi nesyndromickými formami jsou identifikovány 4 skupiny: běžné ichtyózy, autozomálně recesivní vrozené ichtyózy (ARCIs), keratinopatické ichtyózy a další méně časté ichtyózy.Tradičně byla skupina ARCIs rozdělena na 2 poruchy, lamelární ichtyózu (LI) a vrozenou ichtyosiformní erytrodermii (CIE). V nové klasifikaci, harlequin ichthyosis (HI) byl přidán do tohoto group1, protože inaktivujících mutací v genu ABCA12 byly identifikovány jako zodpovědné za tuto poruchu,2,3, zatímco nesmysl mutace ve stejném genu mohou vést k LI4 nebo CIE5,6 fenotyp. Jiné méně běžné varianty zařazené do skupiny ARCIs jsou samoléčivé kolodiové dítě (SHCB), akrální SHCB a ichtyóza plavek.7-9
konsensuální klasifikace založená na klinických rysech Ichtyózy1.
| Nonddromic Formy | Syndromový Formy |
| Společné IchthyosesIchthyosis vulgarisRecessive x-vázaná ichtyóza (nonsyndromic )ajimajor formsHarlequin ichthyosisLamellar ichthyosisCongenital ichthyosiform erythrodermaMinor formsSelf-léčení kolódiové babyAcral self-léčení kolódiové babyBathing oblek ichthyosisKeratinopathic IchthyosesMajor formsEpidermolytic Ichthyosissuperficial epidermolytic ichthyosisminor formsannular epidermolytic ichthyosiscurth-Macklin ichthyosisautosomal recesivní epidermolytic ichthyosisEpidermolytic nevusOther FormsLoricrin keratodermaErythrokeratodermia vararabilispeeling kůže syndromeCongenital retikulární ichthyosiform erythrodermaKLICK syndrom | Syndromový X-vázaná Ichthyosrecessive x-vázaná ichtyóza (syndromový)Ichtyóza follicularis, alopecie, a fotofobie (IFAP) syndromeconradi-Scammann-happle syndrom (chondrodysplázie punctata typ 2)syndromový autosomálně ichthyosisskin disordersnetherton syndromeichthyosis-hypothrichosis syndromeichthyosis-Sklerotizující Cholangitis syndrometrichothystrophyneurological Disorderssjögren-Larsson syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, mentální retardace, enteropatie, hluchota, periferní neuropatie, ichtyóza, keratoderma.
k dispozici jsou pouze omezené údaje o epidemiologii ARCIs. Ve Spojených státech byla odhadnuta prevalence při narození 1 na 100 000 obyvatel pro LI a 1 na 200 000 obyvatel PRO CIE. Jiné studie uváděly kombinovanou prevalenci LI a CIE 1 na 200 000 až 300 000 obyvatel.10,11 v některých zemích, jako je Norsko, je odhadovaná prevalence větší (1 na 91 000) v důsledku mutací zakladatelů.12 zjištění z 1 nebo několika opakujících se mutací v populaci může být, protože došlo k mutaci v daném bodě v historii, a pak byl předán z generace na generaci (zakladatel mutace), nebo proto, že oblasti genomu, kde se mutace je zjištěno, že má DNA sekvence náchylné na mutace (mutace hotspot). Ve Španělsku je odhadovaná prevalence ARCI 1 na 138 000 v běžné populaci a 1 na 61 700 u dětí mladších 10 let.13 v některých regionech Španělska může být prevalence ještě vyšší. Například na galicijském pobřeží byla hlášena prevalence 1 na 33 000, a to také kvůli zakladatelskému efektu.14
Lamelární Ichtyóza a Vrozené Ichthyosiform ErythrodermaClinical Vlastnosti
i když to bylo původně si myslel, že LI a CIE byly různé subjekty, byly hlášeny u pacientů s středně pokročilé klinické projevy a obojí může být způsobena mutacemi ve stejném genu.15,16 kromě toho mohou pacienti se stejnou mutací, dokonce i ve stejné rodině, vyvinout různé fenotypy.12,15
Většina pacientů se rodí zahalen v kolodiu membrána, která postupně zmizí během prvních týdnů života a je nahrazena definitivní fenotyp (Obr. 1A). Hypohidróza, těžká tepelná intolerance a dystrofie nehtů jsou často pozorovány jak U LI, tak u CIE.17-19 pacientů s LI má obvykle závažnější klinické projevy než pacienti s CIE. Mají velké platékové váhy, často tmavé barvy, pokrývající celý povrch těla. Erytrodermie je buď nepřítomná nebo minimální. Tito pacienti mají obvykle ektropium a, občas, eclabium, hypoplazie společné a nosní chrupavky, zjizvení alopecie, zejména na okraji pokožky hlavy, a palmoplantární keratoderma (Obr. 1B a C). CIE se vyznačuje přítomností erytrodermie a jemného bělavého škálování (obr. 2). Někteří pacienti mají výrazný erytém a generalizované škálování. Váhy mohou být velké a tmavě zbarvené, zejména na extenzních plochách nohou. V méně závažných případech je erytém mírný a škálování je v pořádku.

klinické rysy lamelární ichtyózy. A, nahnědlá lamelární deskvamace. B, výrazná plantární hyperkeratóza. C, zjizvená alopecie pokožky hlavy.
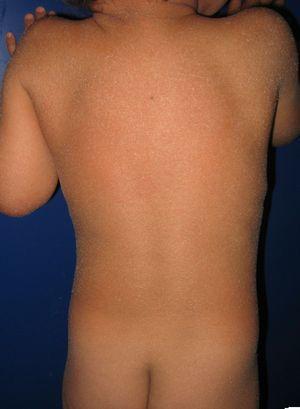
pacient s vrozenou ichtyosiformní erytrodermií a mutacemi v genu ALOXE3. Lze pozorovat mírný erytém a generalizovanou bělavou kožešinovou deskvamaci.
histopatologie
histopatologické změny neposkytují diagnózu. U LI je pozorována masivní orthokeratotická hyperkeratóza, obvykle s dvojnásobným rozšířením než u CIE. Epidermis je akantotická a občas nabývá vzhledu podobného psoriáze. Rychlost proliferace buněk je normální nebo mírně zvýšená.17-19 Pacientů s CIE mají méně výrazný, hyperkeratóza, s fokální nebo rozsáhlé parakeratóza, normální nebo zesílenou granulární vrstva, a výraznější rozšíření. Epidermální obrat se zvyšuje.17-19
Ultrastructure
i když úzká souvislost mezi molekulární, klinické a ultrastrukturální nálezy dosud nebyla nalezena, elektronová mikroskopie přesto mohou být užitečné pro rozhodnutí jiných forem ichtyózy a pro vedení genetické analýzy v některých případech. Byly popsány čtyři typy vrozené ichtyózy(Tabulka 2).
ultrastrukturální klasifikace vrozených Ichtyóz.
| Typ | Hlavní Funkce | Další Funkce | Mutace | Klinické Projevy |
| 1 | Absense ultrastrukturální znaky ichtyóza typy 2, 3, a 4 | Lipidových kapiček nebo kroužky ve stratum corneum (nejčastější)Malé keratohyalin granulesVesicular nebo lobulární membrána povlak granule | TGM1 (33.3%)ALOX12B (2 případy) | CIE |
| 2 | Cholesterol rozštěpů ve stratum corneum | Absence nebo ztenčení zrohovatělé envelopeSmall keratohyalin granulesLipid kapičky | TGM1 (89-100%) | LI |
| 3 | Laminované membraneous struktur ve stratum granulosum a/nebo stratum corneum. | Abnormální membrána povlak granulesLipid dropletsFoci významných juxtanuclear vakuoly v zrnité vrstvě | NIPAL4 (93%) | CIE (nejčastější)LI |
| 4 | Trilamellar membránové pakety, které vyplňují některé buňky ve stratum granulosum a/nebo stratum corneum | Abnormální membrána povlak granule | FTAP4 | Ichtyóza nedonošených syndrom (100%) |
Zkratky: CIE, vrozené ichthyosiform erytrodermie; LI, lamelární ichtyóza.
Vrozená Ichtyóza Typ 1
Vrozená ichtyóza typ 1 je charakterizován absencí ultrastrukturální znaky pro ichtyóza typy 2, 3 a 4. Proto se diagnóza obvykle provádí pouze tehdy, když byly vyloučeny ostatní typy. Nejčastějším nálezem je přítomnost lipidových kapiček nebo kroužků ve stratum corneum (obr. 3A).20 Tyto lipidové kapičky nejsou konstantní funkce nebo specifické pro tento konkrétní typ, protože nejsou přítomny ve všech případech,20 a mohou být přítomny v jiných typech ichtyóza.21,22 klinicky většina pacientů vykazuje projevy CIE.12,20 jedna třetina pacientů má mutace v genu TGM1.16 tento ultrastrukturální typ byl také identifikován ve spojení s mutacemi v genu ALOX12B.23,24

snímky elektronového mikroskopu. A, vrozená ichtyóza typu 1, vykazující lipidové kapičky ve stratum corneum a nepřítomnost ultrastrukturálních markerů jiných typů ichtyózy. B, vrozená ichtyóza typu 2, charakterizovaná přítomností trhlin cholesterolu (šipka) v korneocytech.
Vrozená Ichtyóza Typ 2
Vrozená ichtyóza typ 2 je charakterizován cholesterolu rozštěpů ve stratum corneum (Obr. 3B).21 takové štěrbiny jsou konstantním nálezem u tohoto typu ichtyózy a mohou být detekovány v různých biopsiích u stejného pacienta; léčba perorálními retinoidy nemá na tyto štěrbiny žádný vliv.U některých pacientů s nedostatečnou aktivitou Tgázy 1 bylo také pozorováno 12,25 agregátů hustých elektronů na korneocytech.26-28 klinicky se většina pacientů projevuje závažnými projevy CIE.12 tento ultrastrukturální typ je silně spojen s mutacemi v genu TGM1.12,16
Vrozená Ichtyóza Typ 3
Vrozená ichtyóza typ 3 je charakteristická lamelární membránových struktur ve stratum granulosum a/nebo stratum corneum. Tyto struktury jsou uspořádány v proužcích kolem prázdného prostoru v blízkosti jádra.22,29–31 klinické projevy tohoto typu jsou odlišné od ostatních; nástup ichtyóza je variabilní, olupování a zarudnutí kůže může být nerovnoměrný nebo generalizované, a průhyby jsou zejména ovlivněny. Mutace v genu NIPAL4 jsou zodpovědné za 93% ichtyóz typu 3.32
Vrozená Ichtyóza Typ 4
je Příznačné, že v kongenitální ichtyóza typ 4, některé buňky ve stratum granulosum a stratum corneum jsou vyplněny s trilamellar membrány balíčky.33 Tyto závěry jsou patognomický pro ichtyóza nedonošených syndrom, stav v současné době považován za syndromový forma ichtyózy.34,35
molekulární studie
z genetického hlediska jsou ARCIs velmi heterogenní. Gen TGM1 je spojen s většinou případů, ale byly hlášeny mutace v 5 dalších genech (ALOX12B, ALOXE3, NIPAL4, CYP4F22 a ABCA12). Fischer a kol.36 studoval 520 rodiny s ARCI a identifikovány mutace v alespoň 1 z těchto genů v 78% případů (TGM1 v 32%, NIPAL4 v 16%, ALOX12B v 12%, CYP4F22 v 8%, ALOXE3 v 5%, a ABCA12 v 5%). V jiné studii z 250 pacientů s ARCI různého původu, 38% mělo TGM1 mutace, 6.8% ALOXE3 mutace, a 6,8% ALOX12B mutace.37 V Galicii jsme identifikovali mutace v genech TGM1, ALOX12B, ALOXE3, NIPAL4 a CYP4F22 v 75% studovaných rodin, ale distribuce mutací byla odlišná.14 Gen TGM1 byl mutován v roce 68.7% případů, zatímco Gen ALOXE3 byl mutován pouze u 1 pacienta. Nezjistili jsme mutace v žádném z dalších 3 studovaných genů.
TGM1
Gen TGM1 je umístěn na chromozomu 14q11. 2 a má 15 exonů (GenBank NM-000359.2). Kóduje enzym tgázy 1, což je jeden z enzymů 3 Tgázy, které se nacházejí v epidermis.38 Tento enzym se podílí na tvorbě zrohovatělé obálka katalyzovat vápníku závislé cross-propojení několika proteinů, jako jsou involucrin, loricrin, a prolinu-bohaté na proteiny.39,40 také katalyzuje vazbu ??- hydroxyceramidy ve vnější vrstvě zkorodované obálky s bílkovinami ve vnitřní vrstvě.41,42 u pacientů s mutacemi TGM1 chybí zkorodovaná obálka a aktivita Tgázy 1 je snížena nebo neexistuje.43-47
Od roku 1995, kdy tento gen byl identifikován jako zodpovědný za některé případy ARCI,48-50 více než 110 mutace byly hlášeny u pacientů různého původu. Mutace v TGM1 jsou nejčastější příčinou ARCI.36,37 tato mutace byla nalezena v 55% případů ve Spojených státech a v 84% případů v Norsku.12,51 nejčastější mutací je c.877-2A>G, který byl nalezen u 34% dosud hlášených mutovaných alel.52 vysoká frekvence této mutace v zemích, jako jsou Spojené státy a Norsko, je způsobena zakladatelským efektem.12,53 druhou nejčastější mutací je p. Arg142jeho. Tento a podobné mutace byly hlášeny v zemích, jako je Egypt, Německo, Finsko a Spojené Státy,15,49-51,54-56 a zdá se, že tyto jsou hotspot mutace.57 mutace Arg307Trp je v japonské populaci častá.5 V Galicii, s. Arg760X, c. 1223_1227delACACA a c.984+1G>mutace v TGM1 byly identifikovány v 81.82% rodin s mutací v tomto genu, což naznačuje, zakladatel efekt.14 potvrzení této hypotézy bylo získáno haplotypovou studií (dosud nepublikovaná práce).
TGM1 mutace jsou zodpovědné za většinu případů LI15,27,44,46,56,58-63 a pro malé procento případů CIE.43,47,64,65 takové mutace mohou také vést k dalším formám ARCI, jako je SHCB, akrální SHCB a ichtyóza plavek.
Mnoho studií se pokusil prokázat genotyp-fenotyp asociace mezi mutací v TGM1 a ultrastrukturální nebo klinické nálezy, ale žádné významné korelace byla pozorována na rande.15,16,53 obecně jsou pacienti s mutacemi v genu TGM1 závažněji postiženi než pacienti bez těchto mutací. Ve studii z 83 pacientů s ARCI ve Švédsku a Estonsku, přítomnost ektropium a kolódiové dítě bylo spojeno s TGM1 mutace, zatímco vyšší míra erytém byl pozorován u pacientů bez mutace v tomto genu.66 další studie ukázala, že typ škálování je hlavním rozdílem mezi nosiči a nenosiči mutací tgm1, při zjištění, že všichni pacienti s mutacemi v tomto genu měli lamelární škálování, zatímco 80% pacientů bez mutací TGM1 mělo jemné škálování.14 kromě toho, bylo vidět, že zkrácení mutace jsou častěji spojené s hypohidrosis a pocení poruchy než missense mutace.51 V severoamerické populace, modelu založeného na přítomnosti určitých klinických charakteristik předpovídá, že pacienti, kteří se narodili jako kolódiové děti a mít oční poruchy a/nebo alopecie jsou 4 krát více pravděpodobné, že mají TGM1 mutace.51
ALOXE3 a ALOX12B
ALOXE3 a ALOX12B geny jsou umístěny na chromozomu 17p13.1.67 mají podobnou strukturu s 15 exonů, které kódují epidermální LOXs eLOX-3 a 12R-LOX.68,69 skutečnosti, že jsou převážně vyjádřena v suprabazálních vrstvách epidermis podporuje jejich roli v pokročilých fázích epidermální diferenciace, s účastí na zpracování lamelárními tělísky.24,70 tyto enzymy působí na sousední kroky v cestě hepatilinu (obr. 4). 12R-LOX transformuje kyselinu arachidonovou na kyselinu 12R-hydroxyeikosatetraenovou, zatímco eLOX-3 přeměňuje tento produkt na epoxyalkoholový izomer69, 71 rodiny hepoxilin A3.72 produkt hepoxilinu je nestabilní a je hydrolyzován v buňkách na specifický trihydroxyderivát (trioxilin). I když přesná úloha produkty hepoxilin cesta není známo, to bylo spekuloval, že se mohou podílet na tvorbě mezibuněčné lipidy stratum corneum, nebo působí jako signály pro navození diferenciace keratinocytů.
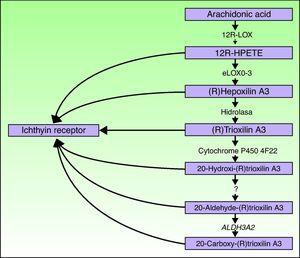
schematický diagram dráhy hepatilinu, ukazující účast genů ALOXE3, ALOX12B, NIPAL4 a CYP4F22. Mutace v těchto genech jsou zodpovědné za některé typy ARCI. HPETE označuje kyselinu hydroperoxyeikosatetraenovou.
Na ALOX12B a ALOXE3 geny byly poprvé identifikovány v 2002.73,74 Od té doby, více než 30 mutací v genu ALOX12B gene23,24,37,75-77 a přibližně 10 v ALOXE3 gene37,74,75 byly hlášeny. Tyto mutace jsou zodpovědné za 14% až 17% ARCIs36,37 a 72.2% Shcb.23,78,79 kauzální vztah mezi těmito mutace a fenotypem byla potvrzena tím, že prokáže, že katalytickou aktivitu epidermální LOX byla zcela zrušena u pacientů s těmito mutations75,80 a pomocí zvířecích modelů, které reprodukoval ichthyosiform fenotyp u člověka.81-83 oba geny jsou zodpovědné za podobné procento případů ARCI. Nicméně, řada různých mutací v genu ALOXE3 je omezen, vzhledem k převaze 2 mutace, p.Arg234X a p.Pro630Leu, které se zdají odpovídat hotspotů.37,74,75
pacienti s mutacemi v genech ALOXE3 a ALOX12B obvykle vykazují fenotyp CIE.74,75,77 závažnost škálování je mírná nebo střední a váhy mají bělavou nebo světle hnědou barvu. Může být také přítomen erytém. Až 76% pacientů se narodilo jako kolodiové děti a 88% má poruchy pocení.37 pacientů s mutacemi v genu ALOX12B vykazuje omezenější bělavou deskvamaci ve srovnání s nosiči mutací v genu ALOXE3. V těchto případech jsou váhy nahnědlé a přilnavé. Přítomnost erytému, palmoplantární hyperkeratózy a zvýraznění palmoplantárních záhybů je také spojena s mutacemi ALOX12B.37
Ichthyin / NIPAL4
Gen NIPAL4, známý také jako gen ichthyinu, se nachází na chromozomu 5q33. Má 6 exonů, které kódují protein s několika transmembránovými doménami neznámé funkce.84 To bylo předpokládal, že proteinový produkt se podílí na stejné metabolické dráhy jako LOSOSEM a může fungovat jako receptor pro trioxilins A3 a B3 nebo pro jiné metabolity hepoxilin metabolické dráhy.84 bylo by tedy zapojeno do tvorby lamelárních těles nebo do jejich transportu směrem k extracelulárnímu prostoru.32 na podporu tohoto jsou 2 pozorování. První, v 93% případů, mutace v tomto genu jsou spojeny s ultrastrukturální vzor vrozená ichtyóza typ 3, vyznačující se tím, abnormalitami v lamelárními tělísky a přítomnost podlouhlých perinukleární membrány ve stratum granulosum.32 druhý, NIPAL4 je exprimován v podstatě ve stratum granulosum epidermis, kde jsou přítomna lamelární těla.85
Od objevu NIPAL4 genu v 2004,84 pouze 9 mutace byly hlášeny u pacientů ze Středomořských zemí (Alžírsko, Turecko a Sýrii),84 Skandinávských zemích,32 Pákistánu,85 Faerské Ostrovy,32 a Jižní Americe.84
klinické spektrum pacientů s mutacemi v tomto genu je široké, dokonce i mezi členy stejné rodiny. Mezi 3,7%32 a 60%84 se rodí jako kolódiové děti. Když kolódiový membrána zmizí, většina pacientů rozvíjet projevy CIE, s jemnými bělavými šupinami na erytematózní základ, na obličeji a trupu a větší, hnědé šupiny na krku, hýždě a nohy.84 může být přítomna výrazná xeróza, generalizované nahnědlé retikulární hyperkeratotické plaky, které se objevují zvýrazněné v kožních záhybech,a dyschromie obličeje.32,85 palmoplantární keratoderma je navíc častým nálezem spolu s občasnými kontrakcemi prstů a zakřivenými nehty prstů. Některé studie uvádějí nálezy typičtější pro LI.32,85 u některých pacientů byla hlášena přítomnost známek a příznaků atopické dermatitidy, ačkoli mutace v genu FLG nebyly v žádném z těchto případů detekovány.85
CYP4F22
FLJ39501 nebo CYP4F22 gen se nachází na chromozomu 19p13.12.86 má 12 exons87 a kóduje cytochromu P450, rodina 4, podčeledi F, polypeptidové 2, homolog leukotrien B4 – ω-hydroxylázy (CYP4F2). Reakci katalyzovanou produktem FLJ39501 v kůži a substráty této reakce lze odvodit analogicky s jejími známými homology CYP4F2 a CYP4F3.88 To bylo předpokládal, že CYP4F2 a CYP4F3 účasti v hepoxilin cestou podle katalyzující konverzi trioxilin A3-20-hydroxy-(R)trioxilin A387 a že konečný produkt této dráhy, 20-karboxy-trioxilin A3, mohou mít základní biologické regulační účinek na kůži.89
K dnešnímu dni, pouze 8 mutace tohoto genu byly hlášeny u 12 příbuzenské rodiny ze Středomoří countries87 a v 1 rodině Izraelského původu.62
v rodinách hlášených Lefèvre et al.,87 většina pacientů měla CIE fenotyp při narození a následně postupoval na LI. Pacienti byli obvykle rodí s vyznačenými erytrodermie, i když bez kolódiové membrány. Jak stárli, vyvinuli generalizované bělavě šedé škálování, které bylo výraznější v periumbilikální oblasti, na hýždích a na spodní části těla. Hyperlinearita dlaní a chodidel a deskvamace na pokožce hlavy, v době pityriasiformního typu, byly časté.87 V jiné rodině, 3 členové postižené narodili jako collidion děti a vyvinula intenzivní erytrodermie, generalizovaná deskvamace, a palmoplantární keratoderma.62
ABCA12
V roce 2003 genu ABCA12 byl ohlásen být zodpovědná za některé případy LI a byl mapován na chromozomu 2q34.4 následně bylo potvrzeno, že mutace v tomto genu byly také zodpovědný za ZDRAVÍM.2, 3ABCA12 kóduje 53 exonů a patří do rodiny ABC transportérů, které váží adenosintrifosfát a zároveň usnadňují transport několika molekul přes buněčnou membránu.90 členové podčeledi ABCA jsou všichni zapojeni do transportu lipidů.91 nedostatečná funkce ABCA12 způsobuje poruchy transportu lipidů v lamelárních tělech a vede tak ke snížení mezibuněčných hladin lipidů ve stratum corneum.3ultrastrukturální studie ukázaly, že ABCA12 se nachází v lamelárních tělech spojených s glykosylceramidy.Mutace 91ABCA12 byly spojeny s poruchami distribuce a transportu glykosylceramidů a se sníženými hladinami hydroxyceramidů, jedné z hlavních složek lipidové bariéry v mezibuněčných prostorech.3,6,92,93 masivní hyperkeratóza, která se vyskytuje u těchto pacientů, by mohla být kompenzační odpovědí na nedostatečnou lipidovou bariéru.94 To by mohlo být také kvůli nedostatku deskvamace corneocytes,93, které by mohly být způsobeny defekty na přepravu určitých proteázy, jako callicrein 5 a cathepsin D, vyplývající z poruch v lamelárními tělísky.95 myších modelů a studie in vitro naznačují, že mutace ABCA12 mají také vliv na epidermální diferenciaci.95-97
K dnešnímu dni více než 50 mutací byly popsány v genu ABCA12 u pacientů s ARCI z Afriky, Evropy, Pákistánu a Japonsku. Nejčastější jsou mutace p.Val244SerfsTer28,2,98,99 identifikovány v Pákistánské a Indické populace, a p.Asn1380Ser,4 identifikovány v Afrických rodin. V obou případech se může jednat o zakládající mutace.
rozsah mutací ABCA12 souvisí s fenotypem, s mutacemi spojenými s úplnou ztrátou funkce vedoucí k HI fenotypu.2,3,98-102 naproti tomu v LI a CIE je většina mutací missense a má méně závažný účinek na funkci bílkovin.4-6, 103 mutace, které jsou základem fenotypu LI, se zdají být koncentrovány v první vazebné kazetové oblasti adenosintrifosfátu.4 Klinicky, pacienti s CIE a mutace v genu ABCA12 mají střední váhy, které jsou poněkud větší než obvykle pozorovány u pacientů s tímto fenotypem.
Harlekýnská ichtyóza
HI nebo harlekýnský plod je těžká a obvykle fatální forma ichtyózy. Děti jsou obvykle předčasně s rozsáhlými lesklé hyperkeratotické plakety, které jsou odděleny hluboké trhliny, které pokrývají celou vrstvou a tvoří geometrické vzory připomínající oblečení, které nosí harlekýny, čímž stav jeho jméno. Pevnost kůže vede k významnému everze víčka a rty, základní rozvoj společných a nosní chrupavky a, občas, mikrocefalie. Děti mají zřídka řasy nebo obočí, i když vlasy na pokožce hlavy mohou být zachovány. Ruce a nohy jsou oteklé a edematózní a často pokryté vrstvou podobnou rukavici. Mohou mít kontraktury prstů.
u těchto pacientů je riziko úmrtí během novorozeneckého období velmi vysoké.104 plicní ventilace je ohrožena; transepidermální ztráta vody vede k dehydrataci, vodní nerovnováze a tepelné nestabilitě; a zvyšuje se riziko infekcí. Těsnost obličeje a eklabium brání sání, a proto krmení, s odpovídajícím zhoršením dehydratace. Novorozenci s tímto stavem zřídka žili déle několik týdnů. V posledních letech se však šance na dlouhodobé přežití výrazně zvýšily, zejména v důsledku podávání systémových retinoidů a pokroku v intenzivní novorozenecké péči.105 v nedávné studii přežilo 83% pacientů léčených perorálními retinoidy ve srovnání s 24% neléčených pacientů. Většina úmrtí nastala v prvních 3 dnech života, ale léčba nebyla zahájena až poté u mnoha přeživších.104 to by naznačovalo, že k mnoha z těchto časných úmrtí by došlo bez ohledu na léčbu retinoidy.
u dětí, které přežijí novorozenecké období, se obecně vyvine závažná CIE.106 povaha a umístění mutací v genu ABCA12 a rozsah ztráty funkce transportéru mohou určit prognózu.3,92,107 Pacientů, kteří zachování určité míry bílkovin činnost, byť minimální, může mít lepší šanci na přežití. Nosiče homozygotních mutací mají vyšší úmrtnost.104
hlavní histologickou charakteristikou HI je přítomnost extrémně silné a kompaktní orthokeratotické stratum corneum. Vlasové folikuly a potní kanálky mají prominentní hyperkeratotické plugs107,108 a mají abnormální nebo chybějící lamelárními tělísky, lipidové inkluze, nebo zbytky organel nebo jádra v corneocytes, a absence mezibuněčných lipidů v ultrastrukturální studie.108,109 vlasové folikuly vykazují výraznou koncentraci keratotického materiálu, což je diagnostický rys HI používaný pro prenatální diagnostiku.
K dnešnímu dni, rychlost detekce mutací v genu ABCA12 u pacientů s HI je téměř 100%, a tak by se zdají být geneticky homogenní stavu.
Kolódiové Dítě a Self-léčení Kolódiové Dítě
Kolódiové děti jsou obvykle rodí předčasně a perinatální morbidity a mortality se zvýšil. Při narození je novorozenec pokryt lesklou průhlednou membránou připomínající celofánový obal (obr. 5). Děti mají ektropion, eklabium a hypoplazii nosní a kloubní chrupavky. Může být zabráněno sání a plicní ventilace110 a transepidermální ztráta vody a riziko infekcí se zvyšuje.110,111

kolodiové dítě, které následně postupovalo do fenotypu lamelární ichtyózy.
Collodion baby je obvyklá prezentace pro HI a CIE. Autosomálně dominantní LI,112,113 Sjögren-Larsson syndrom,110 trichothyodystrophy,114 mladistvých Gaucherovy choroby,110 neutrálních lipidů skladování onemocnění, Conradi-Hünermann-Happle syndrom, Hays-Wells syndrom, a ektodermální dysplasia115 může také občas přítomen jako kolódiové dítě. Membrána spontánně zmizí u 10% až 24% novorozenců, aby ustoupila zcela normální kůži.110,116 V minulosti, tyto případy byly popsány jako LI novorozence,117 ale nejsou označovány jako SHCB.118 Někteří autoři se domnívají, termín self-zlepšení kolódiové ichtyóza, protože mnoho z těchto pacientů, když přezkoumán později v dětství nebo jako dospělí, mají různý stupeň anhidróza a nesnášenlivost tepla a mírné známky ichtyóza, jako je suchá kůže a jemnou deskvamaci, zejména v axillae a krku.78
optická mikroskopie ani ultrastrukturální vyšetření kolodiového dítěte nejsou specifické. Proto je vhodnější odložit biopsii kůže, dokud se nevyvinul definitivní fenotyp.
mutace v genech TGM1,7,119ALOXE3,78 a ALOX12B23,78,79 byly identifikovány u pacientů se SHCB. Mutace ALOX12B jsou nejčastější. V sérii 15 Skandinávských pacientů s SHCB, 67% mělo mutace v genu ALOX12B, 25% v genu ALOXE3, a 8,3% v genu TGM1.U některých pacientů nebylo nalezeno 78 mutací, a proto jsou pravděpodobně zapojeny i další geny. Spekulovalo se, že tyto mutace snižují enzymatickou aktivitu v děloze, ale ne po narození.7 v děloze, kde je hydrostatický tlak vysoký, chelace vodou přeměňuje mutovaný enzym na neaktivní konformaci. Po narození, když tlak klesá, enzym vrátí do své aktivní formy a její aktivita se zvyšuje dostatečně k udržení normální nebo minimálně ovlivněn fenotypem.7
Akrální Self-léčení Kolódiové Dítě
i když kolódiové dítě ovlivňuje celé tělo, případů omezuje na akrálních oblastí byly hlášeny. V roce 1952 Finlay et al.120 hlásil případ kolodiové membrány, která postihla pouze ruce a nohy a která následovala samoléčebný průběh. Nedávno byl hlášen nový případ akrálního SHCB ve spojení s mutacemi genu TGM1.8 není známo, proč jsou tyto léze omezeny na akrální oblasti, i když mohou být v provozu faktory spojené s regulací enzymové aktivity závislou na místě.8
ichtyóza plavek
ichtyóza plavek byla poprvé hlášena jako nezávislá varianta ARCI v roce 2005, ačkoli případy ichtyózy se zvláštním rozšířením byly hlášeny dříve.121-123 bylo zjištěno hlavně u pacientů jihoafrického původu, 9 ačkoli to bylo také hlášeno u jedinců z Evropy a středomořských zemí.124 při narození mají pacienti generalizovanou kolodiovou membránu, která se pak zbaví charakteristické distribuce škálování. Trup, proximální oblasti zbraní, včetně axillae, krk a pokožku hlavy jsou obecně ovlivněny, zatímco centrální část obličeje, končetin, a nadledvin regionu jsou obvykle ušetřeny.9 šupiny jsou velké, lamelární a tmavé barvy. Jemnější deskvamace může nastat v popliteal a antecubital fossae.124,125 dlaně rukou a chodidel nohou mají mírnou difúzní hyperkeratózu, zatímco záda rukou a nohou nevykazují žádné zapojení.
Histopatologické studie postiženou kůži ukazuje, označené hyperkeratóza bez parakeratóza, normální granulární vrstvy, mírné nebo středně závažné akantóza, a mírný lymfocytární infiltrát v horní dermis.9 pozorování elektronové mikroskopie je ve většině případů v souladu s vrozenou ichtyózou typu 2. Nezúčastněná kůže nevykazuje žádné abnormální nálezy.124,125 u zdravé kůže je aktivita Tgázy 1 mírně snížena a obvykle lokalizována v pericelulárních oblastech. V postižené kůži je enzymatická aktivita zbytková a abnormálně lokalizovaná v cytoplazmě.U všech dosud studovaných pacientů s ichtyózou v plavkách bylo v genu TGM1 detekováno 124
mutací.119,124-126 nejčastější mutací je p. Arg315Leu, který byl identifikován u většiny jihoafrických pacientů a mohl by být zakládající mutací. Oji et al.124 naznačuje, že teplota kůže může hrát roli ve vývoji těchto projevů. Pomocí digitální termografie, autoři ukázala silnou korelaci mezi tělesnou teplotu a deskvamace, s nejteplejší oblastí těla, že ty nejvíce postižené. Aufenvenne et al.127 prokázalo snížení optimální teploty pro aktivitu Tgázy 1 u pacientů s ichtyózou plavek. Tento pokles nebyl pozorován u zdravých dobrovolníků nebo u pacientů s generalizovanou LI. Tento pokles teploty by vysvětlovalo fenotyp těchto pacientů. Optimální teplota je 37°C pro normální enzym, ale 31°C pro mutovaný enzym.
léčba
primárním cílem léčby ichtyózy je odstranění škálování a snížení xerózy, aniž by došlo k nadměrnému podráždění (Tabulka 3). Před rozhodnutím o léčbě je třeba vzít v úvahu aspekty, jako je věk a pohlaví pacienta, typ a závažnost onemocnění a rozsah a místo lézí.128
terapeutická strategie u autozomálně recesivních vrozených Ichtyóz.
| Léčebné strategie pro autosomálně recesivní vrozené ichthyoses | |
| Koupání a mechanické odstranění váhy | Koupání s hydrogenuhličitan sodný nebo pšeničný škrob, kukuřičný škrob nebo rýžový škrob; mechanické odstranění váhy (1 nebo 2 krát denně) |
| Lokální léčba (sekvenční) | Močovina obsahující moisturizersKeratinolytics s propylen glycolCombined keratinolytics (propylen glykol, α-hydroxy kyseliny nebo močovina)Keratinolytics v kombinaci s salicylové acidTopical retinoidsIn novorozenců a malých dětí, naneste vozidla bez účinných látek. Vyhnout močoviny, kyseliny salicylové a kyseliny mléčné vzhledem k riziku systémové absorpci |
| Perorální léčba | Perorální retinoidy (acitretin nebo isotretinoin) |
| Další opatření | Follow-up ektropium, které ophthalmologistRegular čištění vnějšího ucha do ucha-hrdlo-nos specialistPhysiotherapy k prevenci kontraktur.Vyhýbání se namáhavé činnosti ve vysokých teplotách temperatureHydrotherapy |
Koupání a Mechanické Odstranění Váhy
Denní koupání se doporučuje u pacientů s ARCI, aby mechanicky odstranit váhy a stopy hydratační krém. To je jednodušší, pokud je pacient ponořen do vody po dobu 15 až 30 minut. Někteří autoři doporučují přidání hydrogenuhličitanu sodného do lázně, aby denaturalizovali keratiny a učinili vodu alkalickou, a tak usnadnili eliminaci váhy.Mezi 129 dalších produktů, které lze přidat, patří pšeničný škrob, kukuřičný škrob nebo rýžový škrob. Koupací oleje nejsou vhodné, protože mohou vést k okluzi s následným rizikem bakteriální proliferace a zhoršení termoregulace.
topická léčba
zvlhčovače a topická keratolytická činidla jsou obvykle první terapeutickou možností. Zlepšují funkci kožní bariéry a usnadňují deskvamaci. Mohou se objevit mírné lokální nežádoucí účinky, jako je přechodný pruritus, podráždění nebo pocit bodání.
chlorid sodný, močovina, acetát vitaminu E, glycerol a vazelína lze použít jako zvlhčovače a maziva. U pacientů s hustou měřítka a označil hyperkeratóza, 1 nebo více keratolytická činidla, jako je α-hydroxy kyseliny (kyselina mléčná a kyselina glykolová),130 salicylová kyselina, N-acetylcystein,131-133 močoviny (>5%),134 a propylenglykol, které mohou být přidány. Používají se také modulátory diferenciace keratinocytů. Patří sem lokální retinoidy (tretinoin, adapalen, tazaroten), 135,136 kalcipotriol, 137 a dexpanthenol.Lokální retinoidy často způsobují podráždění a malé, velmi bolestivé trhliny.137 kromě toho existuje riziko absorpce a teratogenity u fertilních žen, pokud jsou používány příliš rozsáhle.138 pro zvýšení účinnosti keratolytik a zvlhčovačů může být okluzivní obvaz aplikován ve specifických oblastech refrakterních na léčbu.139 aditivního nebo synergického účinku lze také dosáhnout kombinací 2 nebo více keratolytických činidel nebo zvlhčovačů.Léčba 140-142 by měla být optimalizována pro každého jednotlivce, vzhledem k vysoce proměnlivé povaze stavu a citlivosti kůže a rozdílům v reakci na každou léčbu. Optimalizačnímu procesu lze pomoci tím, že se na jedné straně těla zachází odlišně od druhé, aby se umožnilo srovnání. Novorozenci a malé děti by měli být léčeni s vozidlem bez jakékoli aktivní látky, protože kůže je velmi jemná a citlivá, a většina keratolytika nejsou tolerovány. Kromě toho je riziko perkutánní absorpce topických produktů, jako je močovina, kyselina salicylová a kyselina mléčná, větší.143-145
systémová léčba
perorální retinoidy mají keratolytické účinky, které pomáhají eliminovat šupiny a zabraňují nadměrné hyperkeratóze. Jak isotretinoin, tak aromatické retinoidy (acitretin a etretinát) se ukázaly jako účinné při léčbě ARCIs.128,146,147 Acitretin v dávce 0,5 až 1 mg/kg/d je nejrozšířenější droga, zejména u pacientů s LI.148 Pacientů s CIE může mít více kompletní odpověď a v nižších dávkách.
hlavními nežádoucími účinky jsou mukokutánní poruchy, teratogenita, muskuloskeletální poruchy a abnormální lipidový profil a zvýšení transaminázy.149-152 S ohledem na teratogenní účinky, v případě etretinate a acitretin, léky je třeba se vyhnout v průběhu těhotenství, a pacienti by se měli vyvarovat otěhotnění po dobu 3 let po ukončení léčby.151 Isotretinoin má kratší poločas a je zcela vyloučen z organismu po 1 měsíci, a proto může být preferovanou možností u žen, které si přejí otěhotnět.128
monitorování Léčby by mělo zahrnovat laboratorní práce-s test jater a lipidový profil před zahájením léčby, poté na 1 měsíc a každé 3 měsíce po zahájení léčby. V plodné ženy, těhotenský test by měl být proveden do 2 týdnů před zahájením léčby a účinná antikoncepční opatření by měl být používán od 4 týdnů před zahájením léčby až do 3 let poté (v případě acitretin). Pokud je vyžadována dlouhodobá léčba retinoidy, měl by být sledován růst a vývoj kostí. Někteří autoři navrhují provedení kostní studie před léčbou, po níž následuje roční vyšetření.151 nedávné pokyny nedoporučují provádět rutinní radiografii kvůli možným škodlivým účinkům.152 místo toho se doporučují selektivní radiografické studie u pacientů s atypickou bolestí kostí.152
alternativou k systémové léčbě retinoidy je použití léků známých jako blokátory metabolismu kyseliny retinové, které zvyšují endogenní hladiny kyseliny retinové. Jeden takový lék je liarozole, která byla udělena vzácná onemocnění, stav po léčbě LI, CIE, a HI Evropská Agentura pro léčivé Přípravky a USA Food and Drug Administration.153-155 ukázalo se, že tento lék je v klinických studiích účinnější než acitretin a je také lépe snášen a má lepší farmakokinetický profil.154
Další Zdravotní Péči
U pacientů s ektropium, aplikace z umělé slzy a oční maziva a hydrataci pokožky obličeje a tváře, zejména může snížit oční odvolání. Chirurgická korekce je v závažných případech platnou možností, ale obvykle se to musí opakovat o několik let později. Vodoléčba může být prospěšná.156 Pacienti by měli být poučeni, aby se zabránilo namáhavé fyzické aktivity, kdy okolní teplota je vysoká, vzhledem k tomu, že hypohidrosis s sebou nese riziko mrtvice tepla a křeče. Perorální retinoidy mohou zlepšit termoregulaci.157 fyzioterapie je důležitá pro prevenci kontraktury flexe, zejména v případě HI. Pravidelné čištění vnějšího sluchového kanálu specialistou na ušní krk a nos může zabránit hromadění váhy a zabránit tak ztrátě sluchu.
Genetické Poradenství a Prenatální Diagnostika
Pokud je pacient diagnostikován s ichtyóza, on nebo ona by měla být nabídnuta vhodná genetické poradenství v nichž povaha poruchy, režim přenosu, a riziko budoucích projevů v rodině jsou vysvětleny. Prenatální diagnóza může naznačovat, zda je plod ovlivněn, a pokud tomu tak je, lze nabídnout psychologickou přípravu rodiny a očekávat problémy během těhotenství a porodu. Rodiče mohou mít možnost potratu, pokud není k dispozici žádná léčba. Kromě toho, pokud by v budoucnu byla k dispozici genová terapie pro tyto stavy, prenatální diagnostika by umožnila aplikaci této terapie co nejdříve.
více než 20 let byla prenatální diagnóza prováděna odebráním vzorku biopsie kůže plodu a studiem optické mikroskopie, elektronové mikroskopie nebo imunohistochemie.158,159 Tento invazivní postup může být provedena pouze v pozdních fázích těhotenství, mezi týdny 15 a 23 gestace a byla spojena s 1% až 3% rizikem ztráty plodu.160,161 identifikace molekulárních mechanismů dědičných kožních poruch umožnila mnohem dřívější diagnózu založenou na genetických technikách.102,162–164 Fetální DNA získané amniocentéza provádí mezi týdny 15 a 20 nebo odběru choriových klků mezi týdny, 10 a 12. Riziko ztráty plodu těmito technikami je menší než mezi 0, 5% a 1%.165 dalšími neinvazivními metodami ve vývoji jsou analýza DNA fetálních buněk a volné fetální DNA v mateřském oběhu166 stejně jako použití 3-dimenzionálního ultrazvuku.167,168
Preimplantační genetická diagnostika může být také možné v oplodnění in vitro technik, jako, že pouze oplodněné vejce zdarma mutace jsou implantovány do dělohy, čímž se zabrání nutnosti potratu ve většině případů.169
Budoucí Strategie pro Genetickou Léčbu Ichtyózy
Přestože bylo dosaženo významného pokroku v genetické diagnostice ichtyóza, nové strategie jsou také sledovány na těchto onemocnění.170 kůže je nejdostupnějším orgánem pro terapie přenosu genů, a proto jsou tyto techniky minimálně invazivní.171 kůže má však také jedinečné imunologické vlastnosti, které nepodporují dlouhodobou expresi transgenního produktu.172 V LI se procesu přenosu genu ex vivo podařilo obnovit normální expresi TGM1 a korigovat fenotyp kůže transplantované na zadní straně imunosuprimovaných myší.173 174 nedávno byl také obnoven fenotyp kultivovaných keratinocytů od pacientů s HI v důsledku mutací v genu ABCA12.3
střet zájmů
autoři prohlašují, že nemají žádný střet zájmů.