Genetika rozštěp rtu a patra
 Intro/abstractCleft rtu s nebo bez rozštěpu patra je komplexní vrozené anomálie, které mohou být izolované nebo společně s dalšími malformací. Může být také součástí fenotypu genetického syndromu. Tento článek slouží jako přehled prevalence rozštěpu rtu a patra, rizika recidivy, a rizika dalších vrozených anomálií. Budou prozkoumány genetické syndromy a teratogenní expozice, o nichž je známo, že jsou spojeny s orálními rozštěpy. Kromě toho budou diskutovány genetické testy běžně požadované v prostředí pediatrické klinické genetiky pro hodnocení pacienta s rozštěpem rtu a patra.
Intro/abstractCleft rtu s nebo bez rozštěpu patra je komplexní vrozené anomálie, které mohou být izolované nebo společně s dalšími malformací. Může být také součástí fenotypu genetického syndromu. Tento článek slouží jako přehled prevalence rozštěpu rtu a patra, rizika recidivy, a rizika dalších vrozených anomálií. Budou prozkoumány genetické syndromy a teratogenní expozice, o nichž je známo, že jsou spojeny s orálními rozštěpy. Kromě toho budou diskutovány genetické testy běžně požadované v prostředí pediatrické klinické genetiky pro hodnocení pacienta s rozštěpem rtu a patra.
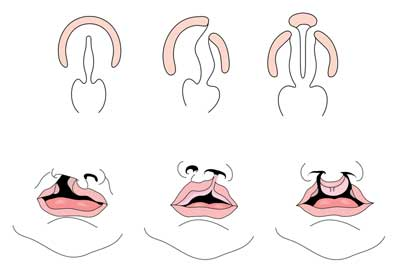
Rozštěp rtu s nebo bez rozštěpu patra (CL/CP) se liší od izolovaného rozštěpu patra (CP) na embryonální, epidemiologické a genetické úrovni. Rozštěp rtu obvykle výsledky z maxilární prominence a mediální nasální výběžek nedaří skloubit mezi pátého a šestého týdne embryonálního vývoje. Normální vývoj patra je výsledkem tvorby primárního patra a sekundárního patra. Primární patro je tvořeno v šesti až sedmi týdnech vývojem a fúzí mediálních nosních, laterálních nosních a maxilárních procesů. Sekundární patro pochází z patrových regálů (které se vyvíjejí z párových čelistní procesy první branchiální oblouk) stává horizontální a tavení, tvarování tvrdého a měkkého patra kolem devátého týdne embryonálního vývoje. Police se také spojují s primárním patrem a nosní přepážkou. (1)
orální rozštěpy jsou jednou z nejčastějších vrozených vad pozorovaných v novorozenecké školce s celkovou prevalencí 1.6 na tisíc novorozenců po celém světě, s CL/CP viděn v přibližně jednou za tisíc porodů a CP viděl v 0,6 na tisíc porodů. (2) u jedinců asijského, afrického a indiánského původu je vyšší frekvence CL/CP. CL / CP je také častější u mužů. V porovnání, neexistuje žádný významný rozdíl ve výskytu CP mezi různými etnickými původy, a CP je častější u žen. (3) rizika recidivy v rodině závisí na tom, zda je rozštěp izolován (bez přítomnosti jiných klinických nálezů) nebo je považován za součást genetického syndromu. Většina případů perorálních rozštěpů je izolována (přibližně 80%). Předpokládá se, že izolované rozštěpy mají multifaktoriální dědičnost: jsou způsobeny kombinací více faktorů, genetických i environmentálních. Riziko recidivy (Tabulka 1) se zvyšuje, pokud existuje více než jeden postižený příbuzný. Riziko recidivy se také zvyšuje, čím závažnější je vada.

rozštěp rtu a patra lze pozorovat u jiných vrozených anomálií. Pravděpodobnost genetické nebo teratogenní etiologie zvyšuje více vrozených anomálií, se kterými pacient představuje. Přítomnost dalších otázek, jako je mentální postižení, problémy s chováním, jako je autismus, poruchy funkce, nebo jiné zdravotní problémy, bude také genetická porucha nebo teratogenní expozice více pravděpodobné. Přibližně 13% jedinců s rozštěpem rtu bude mít jiné zdravotní problémy nebo anomálie. Počet se zvyšuje na 37% s rozštěpem rtu a patra a na 47% pouze s rozštěpem patra.
Prenatální expozice teratogenní látky (jako jsou thalidomid, antikonvulziva, alkohol, kyselina retinová a cigarety) a matky onemocnění (jako je diabetes, zarděnky, a folátu) bylo prokázáno, že zvyšují riziko orálních rozštěpů. Přítomnost amniotických pásů také zvyšuje riziko rozštěpů. Je známo, že perikoncepční suplementace kyselinou listovou snižuje riziko perorálních rozštěpů.
Pierre Robin sekvence je kraniofaciální anomálie se vyznačuje tím, mandibulární hypoplazie nebo mikrognacie, sekundární ve tvaru U rozštěpu patra, a glossoptosis což vede k obstrukční apnoe a krmení problémy. Sekvenci Pierra Robina lze považovat za součást genetických syndromů (deleční syndrom 22q11.2, Sticklerův syndrom; popsáno níže). (5)
Existují stovky genetické syndromy spojené s ústní rozštěpy, včetně cytogenetických abnormalit (aneuploidií, microdeletions) a single-gene (Mendelistická) poruchy. Potvrzení genetické diagnózy je nezbytné pro stanovení prognózy a stanovení rizika recidivy.
aneuploidie, jako je trizomie 13 a 18, mají silnou souvislost s CL / CP. Trizomie 13 (AKA Patauův syndrom), je spojena s tři kopie chromozomu 13, nebo nevyvážené Robertsonské translokace zahrnující chromozom 13. Děti narozené s tímto stavem obvykle umírají v novorozeneckém období. Klinické rysy patří rozštěp rtu a patra, růstové retardace, těžkou centrální nervový malformace (včetně holoprosencefalie), mikrocefalie, micropthalmia, kolobom duhovky, absence očí, znetvořené uši, polydaktylie, zaťaté pěsti, rocker spodní nohy, vrozené srdeční vady a urogenitální vady. U trizomie 13 lze pozorovat rozštěpy střední linie (jinak velmi vzácné) kvůli riziku defektů střední linie, včetně holoprosencefalie. Trizomie 18 (AKA Edwardsův syndrom) je obvykle způsobena třemi odlišnými kopiemi chromozomu 18 a je spojena se špatným postnatálním výsledkem. Klinické rysy patří rozštěp rtu a patra, mentální postižení, porucha růstu, vrozené srdeční choroby, hypertonie, mikrognacie, krátká hrudní kost, nízká nastavit znetvořené uši, zaťaté ruce, rocker spodní nohy, a hypoplastické nehty, mezi ostatními. Trizomii 13 a 18 lze snadno potvrdit nebo vyloučit provedením analýzy chromozomů (karyotypizace).
Mikrodeleční syndromy obvykle zahrnují deleci části chromozomu. Tyto delece mohou být příliš malé na to, aby mohly být detekovány standardní karyotypizací, a mohou vyžadovat detekci FISH (fluorescence in situ hybridizace) nebo technologie microarray. Známý mikrodeleční syndrom spojený s rozštěpem patra je deleční syndrom 22q11.2 (aka Digeorge / Velocardiofaciální syndrom). Patrový abnormality včetně velopharyngeal neschopnost, submukózní rozštěpy, bifid čípek a rozštěp patra jsou vidět v 69% jedinců s 22q11.2 odstranění, a může být součástí Pierre Robin sekvence. Další klinické nálezy patří vrozené onemocnění srdce, ztráta sluchu, poruchy funkce, imunity, hypokalcémie, renální anomálie, krmení problémy, anomálie skeletu, a psychiatrické poruchy. Přibližně 10% případů delečního syndromu 22q11. 2 je považováno za familiární. Odstranění se segreguje autosomálně dominantním způsobem.(6) Wolf-Hirschhornův syndrom, který je způsoben delecí v krátkém rameni chromozomu 4, je také spojen s orálními rozštěpy (u 25% až 50% postižených jedinců). Charakteristické rysy obličeje (včetně předních průčelí vedoucí do “řeckého warrior přilba vzhled”), vrozené srdeční vady, mentální postižení, křeče, neprospívání, mikrognacie, preauricular tagy nebo jámy, a hypodoncie může také být viděn jako součást stavu.(7)
Single-gene poruchy s ústní rozštěpy patří Stickler syndrom, Treacher Collins syndrom, a Van der Woude syndrom, mezi mnoho jiní. Sticklerův syndrom je kolagenová porucha s autozomálně dominantní a méně často autozomálně recesivní dědičností. Společné funkce patří rozštěp patra (viděl jako součást Pierre Robin sekvence, nebo bez mikrognacie), ztráta sluchu (senzorineurální a vodivé), kosterní nálezy (časný nástup artritidy, spondyloepiphyseal dysplazie), oční anomálie (vysoká myopie, skelná abnormality) a charakteristické rysy obličeje (s zaostalosti horní čelisti a nosní most, střední část obličeje retrusion). Genetické testování Sticklerova syndromu může být složité, protože u postižených jedinců byly popsány mutace v nejméně šesti genech. Přibližně 90% pacientů s Sticklerovým syndromem má mutace v genu COL2A1 a má autozomálně dominantní formu stavu.(8) Treacher Collins syndrom je autozomálně dominantní stav charakterizovaný rozštěpem patra s rozštěpem rtu nebo bez něj u 28% postižených jedinců. Jiné abnormality patří hypoplazie lícní kosti a dolní čelist, anomálie zevního ucha, kolobom dolního víčka, vodivé ztráta sluchu, absence spodní řasy, preauricular vlasy posunutí na lících, a choanal stenóza nebo atrézie. Diagnóza syndromu Treacher Collins je založena na klinických a radiografických nálezech. Byly popsány mutace v nejméně třech genech, přičemž mutace v TCOF1 byly pozorovány u 78% až 93% pacientů.(9) Van der Woude syndrom je charakterizován přítomností vrozené, obvykle bilaterální, paramedian spodního rtu píštěle (jámy), nebo někdy malé mohyly s sinus traktu vedoucí ze sliznice žlázy na rtu, a orálních rozštěpů (včetně CL/CP a CP). Van der Woude je autozomálně dominantní stav spojený s mutacemi v genu IRF6 (10). Testování stavu jednoho genu nebo více genů vyžaduje přímou analýzu genu sekvenováním a/nebo delecí / duplikací (jako je MLPA).
Vzhledem k tomu, že genetické syndromy s rozštěpem rtu a patra může být spojen s aneuploidií, chromozom microdeletions/microduplications, nebo single-genové poruchy, genetické testování může být složitý proces. Důkladná anamnéza, třígenerační rodokmen, anamnéza těhotenství a dysmorfologické vyšetření klinickým genetikem mohou objasnit klinický obraz a umožnit cílené genetické testování. Novější technologie včetně microarray umožní identifikaci malých mikrodelecí a mikroduplikací, které dříve standardní karyotypizace chyběly. Bohužel, tato technika také vede k identifikaci delecí a duplikací neznámého klinického významu, což komplikuje genetické poradenství proces. Testování na poruchy jednoho genu nebo Mendelovy poruchy vyžaduje klinickou dostupnost genetického testování požadovaného genu. Může být také drahé, pokud není hrazeno zdravotním pojištěním. Nové technologie, jako je sekvenování nové generace, sekvenování exomu nebo sekvenování genomu (známé souhrnně jako genomické testy), jsou nyní klinicky dostupné. Analýzou stovek až tisíců genů současně tyto testy významně zvyšují diagnostickou sílu a výnos. Ve srovnání s jinými technikami mohou tyto testy poskytnout odpověď rychleji a nákladově efektivnějším způsobem. V oblasti výzkumu vedlo sekvenování exomu a genomu k identifikaci nových genů a rozšíření klinických znaků a spektra genetických mutací. Stejně jako u technologie microarray mohou genomické testy detekovat syndromy, které nesouvisejí s prezentací pacienta a / nebo důvodem testování. Vzhledem ke složitosti genetického testování je nutný informovaný souhlas.
závěr
ačkoli rozštěp rtu a patra je ve většině případů izolovanou anomálií, existuje silná souvislost mezi orálními rozštěpy a jinými anomáliemi a genetickými syndromy. Genetické hodnocení klinickým genetikem a genetickým poradcem je nezbytné pro předvídavé vedení a pro stanovení rizik recidivy. Genetické testování, které vyžaduje informovaný souhlas, lze během genetického hodnocení koordinovat a interpretovat.
Anya Revah, MS, je vedoucí genetická poradkyně v oddělení lékařské genetiky v Maimonides Infants and Children ‘ s Hospital v Brooklynu v New Yorku. Je také aktivní členkou multidisciplinárního týmu Maimonides Medical Center a Kings County Hospital rozštěp rtů a patra. Má magisterský titul v oboru genetického poradenství na Bostonské univerzitě v Bostonu, Massachusetts.
1. Sadler TW. Langmanova lékařská embryologie. Deváté Vydání. Strany 390-395.
2. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer ZNOVU, Anderson, P, Mason CA, Collins JS, Kirby RS, Correa a. Pro Národní vrozené Vady Síť Prevence. Aktualizované národní odhady prevalence narození u vybraných vrozených vad ve Spojených státech. 2004-2006. Výzkum vrozených vad (část a): klinická a molekulární teratologie 2010;88:1008-1016.
3. Fraser FC. Genetika rozštěpu rtu a rozštěpu patra. Rána. J. Hum. Genete. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC, et al. Perikoncepční příjem folátu doplňkem a příjmem potravy snižuje riziko nesyndromického rozštěpu rtu s rozštěpem patra nebo bez něj. Předchozí Med 2004; 39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Vývojové a genetické pohledy na sekvenci Pierra Robina. Rána. J.Med. Genete. 2013; 163C: 295-305.
6. McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11. 2 deleční syndrom. Září. 23, 1999. . In: Pagon RA, Adam MP, Ardinger HH, et al., Editor. GeneReviews . Seattle (WA): University of Washington, Seattle; 1993-2014. Dostupné z: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia A, Carey JC, South ST, et al. Wolf-Hirschhornův Syndrom. Duben. 29, 2002. . In: Pagon RA, Adam MP, Ardinger HH, et al., Editor. GeneReviews . Seattle (WA): University of Washington, Seattle; 1993-2014. Dostupné z: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, Ala-Kokko L.Sticklerův syndrom. Červen. 9, 2000. . In: Pagon RA, Adam MP, Ardinger HH, et al., Editor. GeneReviews . Seattle (WA): University of Washington, Seattle; 1993-2014. Dostupné z: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, popíchnutí fuj. Treacher Collinsův Syndrom. Jula. 20, 2004. . In: Pagon RA, Adam MP, Ardinger HH, et al., Editor. GeneReviews . Seattle (WA): University of Washington, Seattle; 1993-2014. Dostupné z: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy S, et al. Poruchy související s IRF6. Říjen. 30, 2003. . In: Pagon RA, Adam MP, Ardinger HH, et al., Editor. GeneReviews . Seattle (WA): University of Washington, Seattle; 1993-2014. Dostupné z: http://www.ncbi.nlm.nih.gov/books/NBK1407/.