Hranic, v Neurologii
Úvod
Neuroacanthocytosis je nadřazený termín pro skupinu vzácných syndromů, které se vyznačují tím, neurologické příznaky v kombinaci s špičaté deformované červené krvinky (acanthocytes). Tato zpráva se zaměřuje na Chorea-acanthocytosis (ChAc) , což je vzácná onemocnění, onemocnění s odhadovanou 1,000 postižených jednotlivců po celém světě způsobené autozomálně-recesivní mutace v VPS13A (vakuolární třídění bílkovin 13 homolog. A) gen na chromosomu 9q (1). Nemoc probíhá progresivní průběh, příčiny zvýšené úmrtnosti může být odmítnuta motorické funkce korelující s rizikem podmínky, jako jsou dysfagie, ale také náhlé neočekávané úmrtí byly popsány (2, 3). Zatím neexistuje příčinná léčba.
podezřelé soužití acanthocytosis a pohybových poruch, byla poprvé zaznamenána v roce 1970 Levine a Critchley (4, 5) a od té doby byl přijat jako význačný rys tohoto onemocnění. Nicméně, s naší kazuistika popisujeme vzácný klinický fenotyp ChAc chybí zjevné poruchy hybnosti, což naznačuje širší škálu klinických projevů. Diagnostické nástroje musí čelit těmto výzvám, aby stanovily správnou diagnózu, zde navrhujeme molekulární neuroimaging jako klíčovou metodu.
Zpráva
samec index pacienta poprvé představen na 25 let s dva nevyprovokované bilaterální tonicko-klonickými záchvaty. Nebyla tam žádná anamnéza. Vyšetření MRI mozku a EEG v tomto věku byly normální a nebyla zahájena žádná léčba kvůli neochotě pacienta a vzácným příhodám. Pacient však měl pokračující záchvaty, které se nyní projevují jako částečná epilepsie. Popsal opakující se pocity náhlých závratí, které byly diagnostikovány jako vertiginózní aura. Kromě toho, že měl dyscognitive záchvaty, v níž ukázal, buď (a) neodpovídavost a přednášet turecké modlitby, (b) orální automatismy, nebo (c) šmelení s rukou a pronášet zvuky—to vše částečně vyvíjí do tonicko-klonickými záchvaty. Video-EEG nyní ukázalo lokální komplexy špičatých vln v pravém i levém temporálním laloku. V souladu s semiologie záchvatu, diagnóza bilaterální temporální epilepsie byla provedena, a léky s levetiracetamu byla zahájena.
Ve věku 31 záchvaty nebyly zcela potlačeny, jako součást další diagnostiku epilepsie spánkového laloku, pacient podstoupil FDG-PET (Obrázek 1), které prokázalo bilaterální mesiotemporal hypometabolism, v souladu s mesiotemporal záchvat původu. Byl však také výrazný, velmi neobvyklý striatální hypometabolismus, který vyvolal podezření na neurodegenerativní pohybovou poruchu. V důsledku toho bylo zahájeno rozsáhlé následné hodnocení(viz Obrázek 2). Byl proveden FP-cit-SPECT, který přinesl středně bilaterální ztrátu dostupnosti striatálního dopaminového transportéru v souladu s poklesem nigrostriatální integrity (Obrázek 1). MRI s vysokým rozlišením nyní představovala bilaterální atrofii nucleus caudatus (obrázek 3). Neurologické vyšetření ukázalo diskrétní vázaný vzorec chůze a hypomimii, ale žádné jiné postižení extrapyramidového motorického systému a žádné bulbární příznaky, jako je krmení dystonie. Kromě nízkého reflexního stavu na dolních končetinách, u kterého studie nervového vedení potvrdily senzomotorickou axonální polyneuropatii, bylo neurologické vyšetření normální. Neuropsychologické příznaky zahrnovaly aspekty úzkostné poruchy a pacient hlásil potíže s krátkodobou ztrátou paměti. Neuropsychologické testování však nepotvrdilo manifest mnestic poruchy, ale spíše nespecifickou rozptýlení, které by navíc byly posíleny o nedostatečné potlačení záchvatů v té době. Laboratorní vyšetření bylo negativní na jakoukoli symptomatickou epilepsii (tj., protilátky proti autoimunitní encefalitidě, antineuronální protilátky). Mozkomíšní mok nevykazoval žádné známky zánětu, ale mírné zvýšení bílkovin (765 mg / l). Analýza biogenních aminů v mozkomíšním moku odhalila zvýšení glutaminu, které jsme spojili s nedávnými záchvaty. V periferní krvi bylo zjištěno 0, 4% akantocytů. Sérologické testy na Wilsonovu chorobu byly negativní. Patrné bylo přetrvávající zvýšení kreatinkinázy (rozmezí: 1 000-3 000 U / l), což vedlo k podezření na diagnózu mitochondriální poruchy. Pro další vyšetřování byl proveden ischemický laktátový test, který ukázal normální výsledky. Byla provedena svalová biopsie m. gastrocnemicus, ale analýza mitochondriální funkce a respiračního řetězce byla normální.
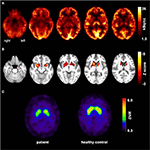
Obrázek 1. Výsledky molekulárních zobrazovacích studií. Transaxiální obrázky FDG-PET (A) ukázaly nejen mírný bilaterální meziotemporální hypometabolismus (v souladu s původem meziotemporálních záchvatů), ale také výrazný striatální hypometabolismus, který byl ve srovnání se zdravými kontrolami vysoce významný . Další FP-CIT-SPECT vyšetření (C) také odhalila mírný úbytek dopaminových transportérů, svědky poklesu nigrostriatální integrity (věk-uzavřeno zdravých kontrol je uveden pro srovnání, DVR, distribuční objem poměr).

Obrázek 2. Klinický a diagnostický průběh indexu pacienta.
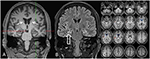
obrázek 3. MRI ukazuje diskrétní bilaterální caudatus hlavy atrofie a menší a hyperintenzivní pravostrannou hipokampu, což naznačuje, pravostranný hipokampální skleróza . Levý hippocampus je poněkud hyperintenzivní, ale ne atrofický. Bilaterální atrofie hlavy kaudátu je potvrzena analýzou CVR, ve které modré barvy vykazují snížené šedé a červené barvy zvýšený objem mozkomíšního moku (C).
rodinná anamnéza odhalila příbuznost jeho rodičů (prvních bratranců), kteří sami neměli žádnou významnou anamnézu. Z jeho šesti sourozenců dva (ve věku 20-30 let) již také trpěli obecnými záchvaty (viz obrázek 4). Lékařské záznamy jeho sourozenců nebyly k dispozici.
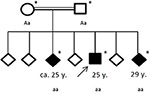
obrázek 4. Rodokmen postižené rodiny s věkem prvního epileptického záchvatu v letech (y). Šipka: index patent. Hvězdička: genetické testování provedeno. Aa, heterozygotní; aa, homozygotní pro c. 4326 T>A (s. Tyr1442*).
podle hypotézy dědičné neurodegenerativní poruchy pohybu byl pacient a jeho rodina odkázáni na genetické testování. Exome sequencing odhalila homozygotní román zkrácení mutace c.4326 T>(str.Tyr1442*) v genu ChAc VPS13A ve tři postižené sourozence. Oba rodiče byli detekováni heterozygotní. Při sekvenování exomu nebyly identifikovány žádné jiné varianty ani mutace.
Ve skutečnosti, v návaznosti na věku 33 let, index pacienta stále jen marginální extrapyramidové příznaky a nikdo nebyl přítomen v každém z jeho sester. Nyní působil dojmem mírné rigidity pouze pod základním nátěrem na pravé horní končetině a mírnou bradykinezí horních končetin. Při léčbě levetiracetamem a lacosamidem byl pacient bez záchvatů.
diskuse
v tomto případě prokazujeme, že ChAc se může klinicky projevit epilepsií temporálního laloku bez významných motorických symptomů v důsledku nové odpovídající mutace v genu VPS13A.
průběh onemocnění je obvykle charakterizována progresivní poruchy hybnosti (včetně chorea, dystonie, parkinsonismus), kognitivní a psychiatrické změny a myopatické příznaky s sérologické markery acanthocytosis a hyperCKemia. Záchvaty byly dříve zaznamenány jako příznak ChAc (6), ale poruchy pohybu jsou stále považovány za klíčový příznak. Existují důkazy, že epilepsie, a konkrétněji epilepsie pocházející z temporálního laloku, může být podceňovaným fenotypem ChAc. Peluso et al. hlásil, že pacient podobné našim, kteří, a to i ve věku 46 let, nevykazují poruchy hybnosti při prezentaci neurologický zjištění orientační bazální ganglia zapojení (7). V souladu s tím, Scheid et al. popsali tři pacienty, kteří trpěli epilepsií meziálního temporálního laloku a sklerózou (na základě studií MRI) jako převládající symptom a byli diagnostikováni s ChAc (8). Dále Mente et al. nedávno poskytl výsledky pitvy pacienta ChAc, který vykazoval nejen atrofii bazálních ganglií, ale také hipokampální sklerózu (9). V souladu s naším případem se bilaterální zapojení hipokampu jeví jako běžné zjištění (10, 11). Přesný vztah mezi záchvaty a sklerózou je však stále otázkou slepice nebo vejce. Korelační role choreinu ve strukturálním vývoji hipokampu není dosud plně pochopena. Je zajímavé, že v myším modelu, Že strukturální změna byla vidět, jako hippokampu expresi bílkovin, GABA (A) receptor gamma 2 a jeho ukotvení proteinu chorein byly zvýšené (12).
v klinické rutině může být tento fenotyp stále náročný při hledání správné diagnózy. První zásadní nápovědu k neurodegenerativní onemocnění v naší pacient byl významný pokles v striatální metabolismus glukózy a mírný pokles v nigrostriatální integrity na FDG-PET a FP-CIT-SPECT, resp. Malá sbírka funkční zobrazování výsledků, na základě případě série popis, ukazují, že změny v metabolismu a dopaminergní dysfunkce se vyskytují hlavně v nucleus caudatus a putamen (13). MRI mozku odhalila atrofii nucleus caudatus v průběhu onemocnění, která byla popsána jako typický nález v ChAc (14). Proto, různé neurologické funkce obvykle patří chorea, krmení dystonie, orofaciolingual dyskineze, a tiky, spolu s dalšími příznaky bazální ganglia náklonnost jako parkinsonismu a dystonie (3). Je lákavé spekulovat, že pozorované striatální změny u našeho pacienta byly stále pod prahem způsobujících symptomů (tj. Proto podporujeme strukturální a molekulární zobrazování jako citlivý diagnostický nástroj u tohoto onemocnění pro vzácná onemocnění.
V ChAc, tam jsou popisy acanthocyte počítá mezi 5 a 50% (3), ale tam jsou některé kazuistiky, které ukazují, že acanthocytes se může objevit pouze pozdě v průběhu onemocnění (15), může být velmi nízké, nebo dokonce zcela chybí (16). Proto by akantocyty neměly být považovány za povinné pro stanovení diagnózy. Navíc epilepsie může oddálit diagnózu, protože maskuje další typické příznaky ChAc. Zejména psychiatrické příznaky, jako jsou změny osobnosti a kognitivní zhoršení, které jsou velmi časté, mohou být nesprávně interpretovány a přičítány vedlejším účinkům souvisejícím s drogami (tj. Sekundární hyperkémie je pozorována po obecných tonických klonických záchvatech a přetrvávající zvýšení může být přehlédnuto.
náš případ také zdůrazňuje zásadní přínos sekvenování exomu pro stanovení správné diagnózy, zejména u vzácných onemocnění nebo pokud jsou příznaky vágní. Rodina savčích VPS13 (A–D) geny, je rostoucí zájem a mutace byly identifikovány u dalších neurologických onemocnění, jako je autozomálně recesivní Parkinsonovy choroby nebo autozomálně recesivní spinocerebelární ataxie (17). Recesivní mutace v VPS13D způsobují poruchy pohybu v dětství (18). Základní patofyziologie ChAc ještě není plně rozluštěn, ale bylo prokázáno, že VPS13A kóduje protein chorein, který se vyjadřuje všude a v mozku a v obrovské řadě dalších tkání (19). Studie ukazují, že se podílí na signálních drahách, které regulují cytoskeletální architekturu, exocytózu a přežití buněk (20). Další mutace, které bylo popsáno prezentovat se záchvat dominuje fenotyp u 9 pacientů s ChAc je c.2343del mutace v VPS13A gen (21). Je rozumné předpokládat, že specifické mutace ve stejném genu vedou k určité aberaci choreinu, která způsobuje jedinečný fenotyp, například ovlivněním různých oblastí mozku. Základní patofyziologie je však stále pouze spekulativní a další dokumentace příčinných mutací bude zajímavá. Ukážeme zde, že zkrácení mutace c.4326 T>(str.Tyr1442*), který nebyl popsáno výše, výsledky v podobným fenotypem s převahou epilepsie u všech postižených členů rodiny.
závěrečné poznámky
epilepsie (zejména bilaterální epilepsie temporálního laloku) je pravděpodobně převládajícím fenotypem ChAc. Proto by mělo být pečlivě provedeno hledání dalších červených vlajek (jako je hyperkémie, rodinná anamnéza, polyneuropatie). V podezřelých případech by mělo být přidáno hledání akantocytů v krvi a MRI mozku, protože tyto nástroje jsou široce dostupné. V případě pochybností by diagnostika měla být doplněna o FDG-PET a / nebo FP-CIT-SPECT, protože jsou citlivé na detekci striatálního postižení. U autosomálně recesivních epilepsií s indikativními nálezy pro neurodegenerativní onemocnění VPS13A by měl být pro stanovení správné diagnózy použit test jednoho genu nebo chorein Western blot. Ta by měla být také zvažována u jiných neurodegenerativních poruch bez geneticky definované etiologie.
etické prohlášení
od pacienta byl získán písemný informovaný souhlas s účastí ve studii. Od pacienta byl získán písemný informovaný souhlas se zveřejněním této kazuistiky.
autorské příspěvky
JW, MR a SK se podílely na řízení pacientů. JW napsal první návrh rukopisu. LF, HU, a PM připravené údaje. Všichni autoři přispěli k revizi rukopisu, přečetli a schválili předloženou verzi.
Prohlášení o střetu zájmů
HU je akcionářem Veobrain Gmbh, spin-off univerzitního Lékařského centra Freiburg.
zbývající autoři prohlašují, že výzkum byl proveden bez jakýchkoli obchodních nebo finančních vztahů, které by mohly být vykládány jako potenciální střet zájmů.
1. Walterfang M, Evans A, Leong Looi JC, Jung HH, Daněk A, Walker RH, et al. Neuropsychiatrie neuroakanthocytózových syndromů. Neurosci Biobehav Rev. (2011) 35: 1275-83. doi: 10.1016 / j. neubiorev.2011.01.001
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
2. Walker RH. Léčba neuroakanthocytózových syndromů. Třes Jiné Hyperkinet. Pohyb. (2015) 5:346. doi: 10.7916 / D8W66K48
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
3. Velayos Baeza A, Dobson-Stone C, Rampoldi L, Bader B, Walker RH, Daněk a, et al. Chorea-Akantokytóza. GeneReviews®. Seattle, WA: University of Washington (1993).
4. Critchley EM, Clark DB, Wikler a. Akantocytóza a neurologická porucha bez Betalipoproteinémie. Arch Neurol. (1968) 18:134–40.
PubMed Abstrakt
5. Levine IM, Estes JW, Looney JM. Dědičné neurologické onemocnění s akantokytózou. Nový syndrom. Arch Neurol. (1968) 19:403–9.
PubMed Abstrakt / Google Scholar
6. Hardie RJ, Pullon HW, Harding AE, Owen JS, Pires M, Daniels GL, et al. Neuroakanthocytóza. klinická, hematologická a patologická studie 19 případů. Mozek (1991) 114(Pt 1A): 13-49.
PubMed Abstrakt / Google Scholar
7. Peluso S, Bilo L, Esposito M, Antenora A, Rosa ADR, Pappata S, et al. Chorea-akanthocytóza bez chorea: rozšíření klinického fenotypu. Parkinsonův Vztah. (2017) 41:124–6. doi: 10.2016 / j. parkreldis.2017.05.013
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
8. Oddělené R, Bader B, Ott DV, Merkenschlager, Daněk, a. Vývoj meziální temporální epilepsie v chorea-acanthocytosis. Neurologie (2009) 73: 1419-22. doi: 10.1212 / WNL.0b013e3181bd80d4
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
9. Mente K, Kim SA, Grunseich C, Hefti MM, Crary JF, Daněk a, et al. Hipokampální skleróza a epilepsie meziálního temporálního laloku u chorea-akantokytózy: případ s klinickým, patologickým a genetickým hodnocením. Neuropatol Appl Neurobiol. (2017) 43:542–6. doi: 10.1111 / nan.12403
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert A, la Fougère C, Noachtar S, et al. Bilaterální epilepsie temporálního laloku potvrzena intrakraniálním EEG v chorea-akantokytóze. Záchvat (2011) 20: 340-2. doi: 10.1016 / j. záchvat.2010.12.007
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
11. Marson AM, Bucciantini E, Gentile E, Geda C. Neuroacanthocytóza: klinické, radiologické a neurofyziologické nálezy v italské rodině. Neurolog. (2003) 24:188–9. doi: 10.1007 / s10072-003-0123-1
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
12. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, et al. Nedostatek choreinu vede k upregulaci gefyrinu a GABA(a) receptoru. Biochem Biophys Res Commun. (2006) 351:438–42. doi: 10.1016 / j. bbrc.2006.10.070
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
13. Ehrlich DJ, Walker RH. Funkční neuroimaging a chorea: systematický přehled. J Clin Mov Disord. (2017) 4:8. doi: 10.1186/s40734-017-0056-0
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
14. Connolly BS, Hazrati L-N, Lang AE. Neuropatologické nálezy u chorea-akantokytózy: nové poznatky o mechanismech, které jsou základem parkinsonismu a záchvatů. Acta Neuropatol. (2014) 127:613–5. doi: 10.1007/s00401-013-1241-3
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
15. Sorrentino G, De Renzo A, Miniello S, Nori O, Bonavita v. pozdní výskyt akantocytů v průběhu chorea-akantocytózy. J. (1999) 163:175–8.
PubMed Abstrakt / Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Choréo-acanthocytóza bez akantocytů. Revue Neurol. (2010) 166:100–3. doi: 10.1016 / j. neurol.2009.03.005
CrossRef Plný Text / Google Scholar
17. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas a, et al. Ztráta funkce VPS13C při autozomálně recesivním parkinsonismu způsobuje mitochondriální dysfunkci a zvyšuje MITOFAGII závislou na PINK1 / Parkinsovi. Am J Hum Genet. (2016) 98:500–13. doi: 10.1016 / j. ajhg.2016.01.014
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
18. Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M, et al. Recesivní mutace u >VPS13D způsobují poruchy pohybu v dětství. Ann Neurolová. (2018) 83: I6. DOI: 10.1002 / ana.25204
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
19. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, et al. In vivo distribuce a lokalizace choreinu. Biochem Biophys Res Commun. (2007) 353:431–5. doi: 10.1016 / j. bbrc.2006.12.059
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
20. Lang F, Pelzl L, Schöls L, Hermann A, Föller M, Schäffer TE, et al. Neurony, erytrocyty a další-různé funkce choreinu. Neurosignals (2017) 25: 117-26. doi: 10.1159/000485457
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
21. Benninger F, Afawi Z, Korczyn AD, Oliver KL, Pendziwiat M, Nakamura M, et al. Záchvaty jako prezentující a prominentní příznak u chorea-akantokytózy s mutací genu c. 2343del VPS13A. Epilepsie (2016) 57: 549-56. doi: 10.1111 / epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar