Hranic, v Pediatrii
Úvod
Familiární nebo hereditární srdeční arytmie tvoří významné procento arytmií a také příčinnou náhlé srdeční smrt (SCD), (1, 2). Během posledních dvou desetiletí vědci a lékaři poskytli obrovské úsilí k odhalení složitých a složitých mechanismů vrozených familiárních arytmií(1-8). Abychom porozuměli mechanismu arytmogeneze, musíme znát základy srdeční buněčné struktury a jejich elektrofyziologické vlastnosti. Srdeční myocyty jsou hlavními fungujícími buňkami v srdci a jsou značně spojeny, takže impulsy se šíří rychle a rovnoměrně. Kardiomyocyty jsou od sebe odděleny speciální hranice, tzv. interkalární disk; gap junction proteiny, srdeční desmozomy a iontové kanály jsou umístěny v interkalární disk (9). Gap junctions se skládají z pevně zabalené connexins, které umožňují mezibuněčnou výměnu malých molekul a také povolení k toku excitační proudy z jedné buňky do sousedních buněk. Desmozomy spolu s adherens junctions jsou zodpovědná za mechanickou příloh jednotlivých kardiomyocytů. Všechny tyto komponenty v interkalárních disků jsou postupně odděleny a každá složka uplatňuje své jedinečné funkce; narušení jedné složky má vliv na funkce ostatních komponent, což předurčuje srdce s arytmií (9-11). Srdeční iontové kanály jsou pórů-tvoří proteinové komplexy, které poskytují napěťových a složitě koordinované aktivního a pasivního pohybu iontové proudy přes buněčné membrány, nezbytné pro srdeční rytmus generace a šíření. Syndrom dlouhého QT (LQTS), krátké syndromem dlouhého QT (SQTS), sick sinus syndrom (SSS), srdeční vedení vada (CCD), BrS, katecholaminergní polymorfní komorová tachykardie (CPVT), časné repolarizace syndrom (ERS), a familiární Fibrilace Síní (AF) jsou v současné době známé srdeční channelopathies, které by mohly nastat v důsledku jednotlivé nebo vícenásobné vady v genech spojena s srdeční rytmus generace a šíření.
v tomto přehledu popíšeme výhradně arytmie spojené s LQTS, její patofyziologii a v současné době dostupné klinické řízení. Jsme průkopníkem v objasnění genetické patologie v rodinném srdeční arytmie v Saúdská Arábie (1, 12-14), stále vedeme cardiogenetic vyšetřování v Saúdské Arábii, na konci tohoto přezkumu, budeme diskutovat v současné době jsou k dispozici znalosti o genetické a klinické nálezy získané ze saúdskoarabské rodiny s anamnézou synkopy a SCD. Přidáme také nedávno zveřejněné údaje o LQTS od týmu v King Faisal Specialist Hospital and Research Center v Rijádu (15).
syndrom dlouhého QT
vrozené LQTS je dědičná porucha definovaná prodloužením QT intervalu na elektrokardiogramu (EKG). Pacienti se všemi formami LQTS jsou náchylní k komorové tachyarytmii, torsades de pointes (TdP) vedoucí k rekurentní synkopě nebo SCD. V mnoha případech může být synkopa nebo náhlá smrt prvním a jediným projevem. LQTS postihuje odhadem 1 z 2 000 lidí na celém světě (16). Charakteristickým znakem LQTS je prodloužení QT intervalu na EKG (korigováno na srdeční frekvenci, tj. Normální hodnoty QTc jsou 440 ms u mužů a 450 ms u žen. U dětí jsou relevantní hodnoty závislé na věku a pohlaví. Nedávné konsensuální doporučení pro diagnostiku LQTS jsou následující (17, 18):
1. LQTS je diagnostikována:
. V přítomnosti LQTS rizikové skóre ≥3,5 v nepřítomnosti sekundární příčinu prodloužení QT intervalu a/nebo
b. V přítomnosti jednoznačně patogenní mutace v jednom z genů LQTS nebo
c. V přítomnosti QT interval korigovaný na srdeční frekvenci pomocí Bazett vzorec (QTc) ≥500 ms v opakované 12-svodového ekg a při absenci sekundární příčinu prodloužení QT intervalu.
2. LQTS může být diagnostikována v přítomnosti QTc mezi 480 a 499 ms v opakované 12-svodového Ekg u pacienta s nevysvětlitelná synkopa při absenci sekundární příčinu prodloužení QT intervalu a při absenci patogenních mutací.
Pacientů s délka QTc ≥500 ms měl výrazně vyšší kumulativní pravděpodobnost výskytu jejich první synkopa než u pacientů s QTc trvání <500 ms od narození do věku 20 let (19). Ale u pacientů, kteří zažili 2, 3 a 4 epizody synkopy, bylo riziko následné epizody synkopy prakticky totožné u pacientů, kteří měli úzké nebo prodloužené trvání QTc (19). Molekulární základ LQTS je heterogenní a dosud byly popsány mutace ve 13 různých genech kauzální k LQTS (1-8 ,19, 20). Genetická vada se obvykle vyskytuje u 70% pacientů s LQTS V jednom z těchto 13 genů (1-8 ,19, 20). Mezi v současné době známo 13 různých typů LQTS, nejčastější jsou LQTS1, LQTS2, a LQTS3 z důvodu vady srdeční iontový kanál genů KCNQ1, KCNH2, a SCN5A, resp. Devadesát procent všech kauzálních mutací LQTS se nachází v těchto třech genech (1-8 ,19, 20). Mutace ve zbývajících 10 genech jsou vzácné a zahrnují pouze ∼10% všech v současné době známých mutací LQTS.
Dlouhý QT syndrom je obvykle autosomálně dominantní onemocnění, ale, občas, více mutací v jediném genu nebo v různé geny lze nalézt v 5-10% pacientů s LQTS (21, 22). Pacienti s více mutací by mohla vykazovat delší QTc intervalu ve srovnání s těmi, s jedinou mutaci, a tito pacienti jsou také v ∼3.5-násobně zvýšené riziko život ohrožujících srdečních příhod (21, 22)
LQTS1
Mutace v genu KCNQ1 jsou nejčastější formu všech LQTS a označován jako LQTS typ 1 (LQTS1). Padesát procent všech kauzálních mutací LQTS se nachází v genu kcnq1 (20). KvLQT1 (také volal Kv7.1) je protein vyrobený podle genu KCNQ1 a bílkovin formy tetramers v endoplazmatického retikula uvnitř buněk, které putatively co-sestavuje s proteinem norek (kódované KCNE1) a jsou poté transportovány k plazmatické membráně srdečních myocytů, kde se zprostředkovat pomalu aktivace proud, který urychluje proces repolarizace akčního potenciálu v srdeční tkání, tento proud je známý jako IKs (Obrázek 1). LQTS1 kauzální mutace KCNQ1 jsou převážně missense mutace a ve vzácných případech mohou být posunovými mutacemi v C-koncové oblasti (23). Srdeční arytmie v nosičích mutací kcnq1 jsou spouštěny adrenergními pohony, např. emoční stres, fyzická námaha, potápění, plavání (24-26). Pacienti s mutacemi v transmembránových doménách KvLQT1 mají vyšší riziko srdečních příhod souvisejících s LQTS a mají větší citlivost na sympatickou stimulaci (26, 27). Pacienti s mutacemi missense jsou vystaveni zvýšenému riziku ve srovnání s pacienty s necitlivými nebo zkrácenými mutacemi (27, 28). Variace v 3 ‘ – UTR v genu kcnq1 také významně ovlivňuje citlivost na arytmii, pravděpodobně ovlivněním exprese genu (29). Pacienti s arytmií v důsledku mutace KCNQ1 reagovat docela dobře na β-blokátory, ale u některých pacientů může být stále méně citlivé, nebo dokonce rezistentní na léky. V nedávném článku, Barsheshet et al. (30) tvrdil, že pacienti s mutacemi mimo oblast cytoplazmatické smyčky (C-smyčky) v KvLQT1 méně reagují na β-blokátory.
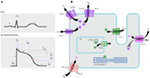
Obrázek 1. Iontové proudy, které přispívají k ventrikulární akční potenciál (a) a schematické znázornění cardiomyocyte zobrazení (pouze) tyto proteiny se podílejí na patogenezi dědičných arytmie syndromy (B). V písmenu a) je akční potenciál vyrovnán s přibližnou dobou působení během EKG. V (B) ankyrin-B, adapter protein podílející se na syndromu dlouhého QT typu 4, není zobrazen.
Homozygotní nebo složené heterozygotní mutace v genu KCNQ1, který by mohl způsobit recesivní forma onemocnění, Jervell a Lange-Nielsen syndrom (JLNS), typ 1 (31), jsou poměrně vzácné. Pacienti s JLNS trpí také těžkými srdečními arytmiemi a hluchotou (31, 32). Pacienti s mutacemi kcnq1 způsobenými JLNS obvykle nemají žádné funkční IKs (28, 31-33). Je možné, že někteří pacienti nemohli mít hluchotu, přestože měli homozygotní nebo složené heterozygotní mutace v KCNQ1, takové případy se označují jako autozomálně recesivní LQTS1 (13, 32). U těchto pacientů, malé množství funkčních IKs proud (<10% z celkového IKs) může být stále přítomen, což udržuje funkce sluchu, ale jejich srdeční rytmus vady jsou stejně závažné jako v JLNS pacientů (13, 32).
LQTS2
Tento typ LQTS je stejně rozšířené jako LQTS1, což představuje 35-40% pacientů s LQTS detekovatelnou mutaci (1, 20, 24, 25). KCNH2 kóduje protein HERG (Kv11. 1), což je α-podjednotka rychle se aktivujícího zpožděného usměrňovače k+ proudu (IKr). Patogenní mutace v tomto genu, které snižují funkci kanálu Kv11. 1, prodlužují trvání QT intervalu (Obrázek 1) a jsou kauzální k LQTS2. Dvacet devět procent synkopálních záchvatů v LQTS2 se vyskytuje během odpočinku / spánku a pouze 13% synkopálních záchvatů bylo hlášeno během cvičení (25, 34). Náhlé překvapivé zvuky, např. zazvoní budík, zvonky, zazvoní telefon, obvykle vyvolávají synkopální útoky u těchto pacientů (24, 34). Pacienti s mutacemi v oblasti tvořící póry Kv11.1 (kódované genem kcnh2) jsou náchylní k vysokému riziku srdečních příhod souvisejících s arytmií ve srovnání s pacienty s mutacemi v oblasti bez pórů (35). U novorozenců je atrioventrikulární (AV) blok 2:1 přednostně spojen s mutacemi kcnh2 (36). Kompletní AV blok komplikovaný LQTS byl také nalezen u 17% dospělých pacientů s mutací v genu kcnh2 (37). Homozygotní mutace v KCNH2 jsou vzácné, a pokud jsou přítomny, pacienti trpí těžkou formou LQTS, s 2:1 AV blok a těžké komorové arytmie, během intrauterinních stadií i po narození (11, 38-40). Kromě četných mutací popsaných v patologii LQTS2 by běžné polymorfní varianty v genu kcnh2 mohly také modulovat závažnost onemocnění. Zajímavým příkladem je polymorfismus K897T (SNP) v KCNH2, který je přítomen u 33% obecné populace (41). Bylo hlášeno, že K897T zhoršuje patogenitu mutací kcnh2 (42, 43).
LQTS3
Nav1.5 je póru tvoří α-podjednotku napěťově závislé srdeční Na+ kanálu, je integrální membránový protein kódovaný genem gen SCN5A a podílí se na zahájení a vedení srdečních akčních potenciálů. Srdeční kanály Na+ se skládají z α-podjednotky tvořící póry (kódované SCN5A) a jedné nebo více pomocných β-podjednotek.
LQTS3 je způsobeno tím, že zisk z funkce mutací, které narušují rychlé inaktivace α-podjednotky (Obr. 1) a mutace v genu SCN5A byly popsány v <10% všech pacientů s LQTS mutaci (1, 20, 24). Pacienti s LQTS3 mají většinu (39%) srdečních příhod během spánku/odpočinku (25) a přibližně ∼13% příhod bylo hlášeno během cvičení (25). V několika případech, jeden SCN5A mutace bylo prokázáno, že vyvinout dva nebo dokonce tři různé fenotypy arytmií ve stejné rodině, např. LQTS, BrS, nebo CCD (44-47). U mužů s mutací LQTS3 se mohou příznaky objevit mnohem dříve než u žen (48). Heterozygotní i homozygotní mutace SCN5A byly popsány v LQTS3 s funkčním AV blokem 2: 1 (49). Nedávno jsme našli íránskou rodinu s mutací 1507_1509delQKP u více členů rodiny, kde pacienti kombinovali LQTS a CCD(údaje nejsou zobrazeny). Tato mutace byla hlášena také u pacientů z jiných zemí, což naznačuje, že se jedná o rekurentní a hot spotovou mutaci . Mutace s takovými charakteristikami ztráty a zesílení funkce během různých fází akčního potenciálu byly také hlášeny u mutací 1493delK a 1795insD (44, 51).
občas by SNP mohl mít také patogenní účinek na své nosiče. S1103Y je běžná varianta v genu SCN5A, přítomná u 13% Afroameričanů (52). Nosiči s touto variantou jsou vystaveni zvýšenému riziku arytmií a syndromu náhlého úmrtí kojenců (SIDS) (52).
LQTS4
LQTS4 představuje první nekanálovou formu LQTS. Mutace v ANK2, adaptér bílkovin, vede k intracelulární vápník přetížení, což přispívá k LQTS4 (53, 54). Kromě prodloužení QT intervalu mohou mít pacienti s tímto syndromem sinusovou bradykardii, paroxysmální AF a CPVT (54). Patogenní účinek ANK2 mutace může být středně těžké až těžké a klinické projevy závisí na závažnosti mutace.
LQTS5
Mutace v KCNE1 jsou spojeny s LQTS5 (Obrázek 1) (55, 56). Heterozygotní mutace kcne1 snižují IKs tím, že vykazují dominantní negativní účinek na doprovodnou normální alelu a vedou ke zpožděné srdeční repolarizaci (Obrázek 1), která je zodpovědná za zvýšené riziko arytmií (6). Pacienti s homozygotními mutacemi kcne1 trpí JLNS (typ 2) (57, 58).
D85N je polymorfismus v genu KCNE1, přítomný v 0.7-1% obecné populace (41). Ve studii Nishio et al. (59), polymorfismus D85N byl častěji nalezen u pacientů s LQTS, což z něj činí rizikový genotyp v patologii LQTS (potenciálně pouze v asijské populaci). V Evropě byl d85n hlášen u 5% pacientů se získanými LQTS (aLQTS) (v kohortě 32 pacientů), kteří měli TdP (60).
LQTS6
Gen kcne2 kóduje peptid 1 související s norky (MiRP1), domnělou β-podjednotku srdečního draslíkového kanálu IKr (Obrázek 1). Mutace v genu kcne2 by také mohly vést k defektům rychle aktivující složky zpožděného usměrňovacího draselného proudu (IKr), patologického základu LQTS6 (61). Sluchový / akustický stimul, jako je hluk budíku, zvonek atd. může vyvolat synkopální útoky v nosičích mutací kcne2, podobně jako mutace kcnh2 (62).
LQTS7
Tento syndrom je také známý jako Andersen–Tawil syndrom (ATS). ATS je vzácná porucha, projevující se občasnou synkopou a srdeční zástavou. Mezi funkce EKG patří mírné prodloužení QT intervalu, abnormální u vlny, častá komorová ektopie, obousměrná komorová tachykardie (VT) a polymorfní VT. Periodická paralýza kosterního svalstva a vývojové problémy, jako je rozštěp patra, nízké uši, krátká postava a vývojové vady končetin (63). U většiny klinicky diagnostikovaných pacientů s ATS byla hlášena mutace v KCNJ2 (63). KCNJ2 kóduje podjednotku tvořící póry vnitřně rektifikujících draslíkových kanálů (IK1) (Obrázek 1) (64, 65).
LQTS8
Také známý jako Timothy syndrom (TS), pacienti vykazují závažné prodloužení QT intervalu na jejich Ekg, který je v kombinaci s syndactyly, plešatost při narození, a malé zuby ve 100% případů a méně kapilární strukturální srdeční vady, mentální retardace, autismus, a obličeje, dysmorfní rysy (66). Existují dva podtypy: TS1 (klasický) a TS2 (vzácná forma).
TS2 je kardiologicky závažnější než TS1 (66, 67). Pacienti s TS2 také postrádají syndactyly (67). Mutace v α-1 podjednotce vápníkového proudu typu L (ICa-L) kódujícího Gen CACNA1C vedou k oběma formám TS (LQTS8). Tam jsou také spojeny, vzájemně se vylučující exon-8 v CACNA1C gen, pro přehlednost jsou pojmenovány jako exon 8 exon-8A. V srdci a mozku, kde CACNA1C je převážně exprimován, exon-8 se nachází v ∼80% mRNA transkriptů a exon-8A je přítomen ∼20% přepisy (66). G406r mutace v exon-8A je kauzální ke klasické formě TS (TS1) a G402S v exon-8 byla hlášena u severera v TS2. Novým přírůstkem do tohoto seznamu je mutace Ala1473Gly, která byla popsána u TS dítěte s dalším rozšířeným fenotypem (68). Všechny mutace byly buď de novo nebo mosaic v mateřské, a všechny výsledek zisk funkce ICa-l kanálu (66-69).
LQTS9 a LQTS10
Mutace v CAV3 nebo SCN4B produkovat zisk funkce v pozdní INa, což způsobuje LQTS3-jako fenotyp (70-74). Jsou známé jako LQTS9 (spojené s mutací CAV3) a LQTS10 (spojené s mutací SCN4B).
Caveolae jsou pěkně popsány Engelmanem et al. (72) jako “malé jeskyně” v plazmatické membráně. Jsou to malé nepotažené jámy a jsou považovány za místo důležitých dynamických a regulačních událostí na plazmatické membráně (72, 73). Caveolins jsou hlavními proteiny v caveolae a caveolin-3 (kódován genem CAV3) je speciálně nalézt v kardiomyocyty a kosterní svalové buňky. Bylo specificky hlášeno, že několik srdečních iontových kanálů je lokalizováno v caveolae extrahovaných ze srdečních myocytů, které jsou obohaceny o caveolin-3 (72, 73). Kromě toho jsou složky signální kaskády β-adrenergních receptorů také přítomny v membránách obohacených o caveolae (72, 73).
SCN4B kóduje pro NaVß4, což je pomocná β-podjednotka srdečního sodíkového kanálu. Dosud byla v tomto genu hlášena pouze jedna mutace (L179F) v mexické rodině s více postiženými členy rodiny (74). Bylo zjištěno, že mutace v tomto genu vede ke zvýšení funkce proudu Nav1. 5 (74).
LQTS11
V srdci, sympatického regulaci srdeční trvání akčního potenciálu (APD) je zprostředkována β-adrenergní receptor (β-AR) aktivace, která vyžaduje shromáždění AKAP9 (Yotiao) s α-podjednotky (KvLQT1) IKs kanálu. Mutace v AKAP9 způsobuje LQTS11 (75). K dnešnímu dni byla hlášena pouze jedna mutace, S1570L, v AKAP9 (75).
Lqts12
mutace v genu α-1-Syntetrofin (SNTN1) jsou kauzální k LQTS12. Mutace v tomto genu vede k získání funkce srdečního sodíkového kanálu (Nav1. 5), který je patologickým základem LQTS12 (76).
LQTS13
G protein-coupled, vnitřně rektifikační podjednotky draslíkového kanálu (Kir3.4) je kódován pomocí KCNJ5 gen. Ztráta funkce mutace v tomto genu by mohla způsobit LQTS13 (77). Dosud byla v čínské rodině popsána pouze jedna mutace, G387R, s devíti pacienty, kteří mají tuto mutaci. Snížená exprese plazmatické membrány Kir3. 4 byla navržena jako patologie LQTS u pacientů.
Získal LQTS
kromě vrozené LQTS, další varianta LQTS známý jako aLQTS také existují, což je způsobeno tím, faktory a látky, které snižují tok draslíku a zhoršit schopnost myokardu se repolarize. Dobře uznávanými stavy jsou ženské pohlaví, hypokalémie a léky, které inhibují srdeční draslíkové kanály (78-80). Řada z běžně předepisovaných léků může také přednostně vázat a blokovat HERG kanál (Kv11.1, protein kódovaný genem genu KCNH2) a předurčuje k aLQTS (79, 80). Nedávno se ukázalo, že blokáda IKs může také přispět k alqt vyvolaným léky, zvláště když je ohrožena repolarizační rezerva (81). Bylo zjištěno, že fluoxetin a norfluoxetin potlačují vlastnosti IKs, a to jak in vivo, tak in vitro a vedly k výraznému LQTS (81). Polymorfismy, D85N v norka (gen KCNE1), T8A, Q9E v MiRP1 (gen KCNE2), které jsou domnělé β-podjednotky z IKs a kanály IKr, byly hlášeny způsobit aLQTS (82, 83). Autoimunitní LQTS byly také hlášeny u pacientů s IgG obsahujícími anti-HERG protilátky (84).
Farmakodynamické Vlastnosti ve Tři Společné Formy Syndromem Dlouhého QT intervalu
Typické ST-T vlny vzory jsou přítomny ve většině genotyp LQTS pacientů a mohou být použity k identifikaci LQTS1, LQTS2, a možná LQTS3 genotypů (85, 86).
forma LQTS1 LQTS je spojena s širokou t-vlnou bez zkrácení QT intervalu při cvičení (obrázek 2A). LQTS2 je spojena s nízkou amplitudou, často bifidními, t vlnami (obrázek 2B). LQTS3 je spojen s dlouhým isoelektrickým segmentem a úzkou, vysokou t vlnou (obrázek 2C). Pauza závislost nástupu TdP u vrozených LQTS je specifická pro genotyp, převažuje v LQTS2, ale téměř chybí v LQTS1 (87).

Obrázek 2. Elektrokardiogramové záznamy od pacientů s LQTS1, LQTS2 a LQTS3. A) 12-olověné EKG 18letého muže s mutací kcnq1. QT interval je prodloužen (QTc = ±500 ms). Segment ST má širokou základnu a relativně velkou amplitudu. Interval vedení je normální (standardní kalibrace). B) 12-olověné EKG 14leté dívky s mutací kcnh2. QT interval je prodloužen (QTc ± 520 ms). Segment ST je vroubkovaný ve vedení V3 a má relativně nízkou amplitudu v koncových vodičích. Interval vedení je normální (standardní kalibrace). C) 12-olověné EKG 12letého chlapce s mutací SCN5A. QT interval je prodloužen (QTc ± 600 ms). Segment ST má dlouhý (téměř) izoelektrický segment s velkou, ostrou a úzkou vlnou T. Interval vedení je normální (standardní kalibrace).
ačkoli vzory mohou naznačovat specifický genotyp LQTS, výjimky byly často nalezeny.
genotyp-fenotyp
syndrom dlouhého QT je autozomálně dominantní onemocnění. Genotyp a fenotyp analýzy mezi heterozygotní nositelé mutace byly provedeny poměrně rozsáhle v LQTS1, LQTS2, a LQTS3 pacientů. Během dětství je riziko srdečních příhod významně vyšší u mužů LQTS1 než u žen LQTS1, zatímco u pacientů s LQTS2 a LQTS3 nebyly pozorovány žádné významné rozdíly v riziku srdečních příhod (88-91). Během dospělosti (také po 40 letech) by ženy LQTS1 a LQTS2 mohly mít významně vyšší riziko srdečních příhod než příslušní muži (88-91). Obecně se zdá, že letalita srdečních příhod převládá u pacientů s LQTS3 než u pacientů s LQTS1 a LQTS2 (91). Ženy s LQTS mají snížené riziko srdečních příhod v průběhu těhotenství, ale riziko se velmi zvyšuje v průběhu 9 měsíců po porodu období, speciálně u žen s mutací v genu KCNH2 (92).
náhlá srdeční smrt u dětí může být také způsobena mutacemi v genech srdečního iontového kanálu (93-96). Asi 28% dětí s nevysvětlitelným SCD (mezi 1 a 18 lety, průměrný věk: 12,3 ± 3,8 let) bylo nositeli mutací v kauzálních genech LQTS (97). V SID, mutace v SCN5A zdálo, že převažují (98, 99), ale mutace KCNQ1, KCNH2, KCNE2, a CAV3, SCN4B, a SCN3B byly také nalezeny (100, 101). Intrauterinní úmrtí plodu byla také hlášena v důsledku defektů v genech srdečního iontového kanálu (12, 102).
Klinické Řízení LQTS
Ukončení všech léků, které jsou známo, že prodlužují QT interval, a také korekce elektrolytové nerovnováhy a/nebo urychlovat metabolické podmínky by měly být hlavním cílem při léčbě (získané-) LQTS pacientů. Symptomy v LQTS jsou často adrenergně zprostředkovány ,proto se obecně doporučuje omezení účasti pacientů na atletických aktivitách (24, 25). Základem klinické terapie pro LQTS je β-blokáda. Dlouhodobě působící přípravky propranolol, nadolol a metoprolol jsou obvykle používány, a jejich účinnost v β-blokátorů je hodnocena otupení cvičení srdeční frekvence (např. >20%) (24, 25). Ze všech β-blokátorů jsou propranolol a nadolol u symptomatických pacientů považovány za lepší než metoprolol (103). Kromě toho může být β-blokáda také použita jako profylaktická léčba u tichých nosičů mutací ke snížení SCD (24). Ve studii Barsheshet et al. (30), pacienti s mutacemi C-smyčky missense v genu kcnq1 vykazovali vysoké riziko život ohrožujících srdečních příhod a měli významný přínos z léčby β-blokátory. Protože ženy s LQTS2 mají zvýšené riziko během 9měsíčního poporodního období, měly by být předepsány β-blokátory ke snížení jakýchkoli srdečních příhod během tohoto vysoce rizikového období (92). Implantovatelné kardioverter-defibrilátory (ICD) lze zvážit u pacientů s rekurentní synkopou navzdory terapii β-blokátory nebo u pacientů s vysokým rizikem srdeční zástavy (např., symptomatické LQTS2 a LQTS3 s dokumentovaným prodloužením QTc). Levé srdeční sympatické denervation (LCSD) je doporučena u vysoce rizikových pacientů s LQTS v níž ICD je kontraindikována nebo odmítnuta a β-blokátory jsou buď nejsou efektivní, nejsou tolerovány, není přijat nebo kontraindikováno (18, 104). Pacienti s JLNS mají obvykle QTc >500 ms a jsou také vystaveni vysokému riziku, β-blokátory mají nedostatečnou účinnost u pacientů, u kterých byla doporučena časná léčba ICD(18, 31).
LQTS: Saúdská perspektiva
první zpráva o LQTS ze Saúdské Arábie byla zveřejněna v roce 1993 z nemocnice Rijádských ozbrojených sil (105). Čtyři kojenci a malé děti ve věku 6 až 48 měsíců s anamnézou opakujících se záchvatů z jedné rodiny byly diagnostikovány jako LQTS (105). Rodinná anamnéza odhalila, že dva další členové rozšířené rodiny měli podobné epizody náhlé ztráty vědomí a tři členové rodiny náhle zemřeli (105). Ve všech případech byla počáteční diagnózou epilepsie (105). O několik let později byly hlášeny dvě sporadické kazuistiky s poměrně závažnější variantou novorozeneckých LQTS v kombinaci s AV blokem 2:1 (106, 107). Všechny tyto publikované klinické zprávy byly bez genetických nálezů, které by mohly vysvětlit patofyziologii LQTS v nich (105-107).
poprvé jsme hlásili genetické defekty jako patologický základ LQTS u podobných pacientů ze Saúdské Arábie. Zkoumali jsme šest saúdských rodin s anamnézou synkopy a náhlými nevysvětlitelnými úmrtími plodu, novorozenců a dětí (12-14). Autozomálně recesivní LQTS1 byl diagnostikován u dětí ze dvou rodin (obrázek 3). Autozomálně recesivní LQTS2 byla diagnostikována ve dvou rodinách (obrázek 4). V jedné rodině, jedné pacientky byla diagnostikována autosomálně dominantní LQTS2 (Obrázek 5), pacient byl v bezvědomí útoky během poporodní rekonvalescence v nemocnici, což je velmi časté v KCNH2 mutace dopravce ženy. Ve všech našich pacientů, identifikace patogenní mutace v LQTS kauzální srdeční iontový kanál genů vedlo k potvrzena klinická diagnóza, které byly misdiagnosed jako epileptické křeče předtím, než je podle nás (13, 14) v Poslední době, Shinwari et al. (15) z King Faisal Specialist Hospital and Research Centre hlášeny LQTS1 kauzální mutace KCNQ1, H258P, ve velké rodině s 12 postižených jedinců. Pouze dva dopravci byly symptomatické, jejich prodloužení QTc >500 ms, a β-blokátory potlačené klinické příznaky u jednoho pacienta, a druhý symptomatický pacient vyžaduje ICD.
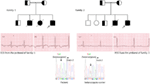
obrázek 3. Rodokmen, kresba rodiny-1 a 2 s autozomálně recesivní LQTS1, probands jsou zobrazeny s šipkou. EKG z probandů ve dvou rodinách jsou zobrazeny uprostřed, s QTc 557 a 529 ms. Intronická mutace, c. 387 -5 t > A v genu KCNQ1 byla nalezena u pacientů (zobrazeno dole). Mutace byla zobrazena šipkou. Nevyplněné kruhy a čtverce nejsou nositeli mutace. Postižení jedinci jsou zobrazeni jako naplněné kruhy (ženy) a čtverce (muži). Napůl vyplněné čtverce a kruhy jsou jedinci s heterozygotní mutací. Zemřelí jedinci jsou označeni lomítky, probandové jsou označeni šipkou a příbuzný sňatek je označen = Exon – hranice intronu je znázorněna tečkovanou čarou se šipkou směřující k exonu.
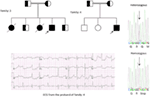
obrázek 4. Rodokmenová kresba rodiny-3 a 4 s autozomálně recesivní LQTS2. EKG probanda od rodiny-4 je uvedeno níže rodokmen výkres, který ukazuje, sinusová tachykardie, téměř kompletní AV blok, široký komplex escape rytmus s velmi dlouhou QTc intervalu (QT >600 ms). Mutace, c. 3208 C > T (p. Q1070X) v genu kcnh2 byla nalezena u pacientů (zobrazených vpravo), označených šipkou.
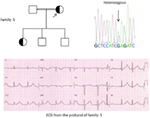
obrázek 5. Vlevo nahoře: rodokmen rodiny-5. Postižený jedinec je zobrazen vyplněným kruhem (samice) Proband je označen šipkou. 12-olověné EKG probandu (I: 2). EKG ukazuje vysoké špičaté, široce založené, velké amplitudy vlny T, prodloužení QTc intervalu je 580 ms. Screening genu KCNH2 ukazuje substituce nukleotidů “G” pro “A” (c.2362G >, šipka označená), která vede k aminokyselinové substituce, p.E788K.
Genetické a klinické nálezy v naší studii (12-14) ze Saúdské Arábie jsou velmi zajímavé z několika důvodů: (1) celkem jsme zkoumali šest rodin, mezi nimi čtyři byli homozygotní/složené heterozygotní mutace a mutace pochází z rodové zdroj; (2) Všechny mutace v LQT byly nové, hlášené pouze v těchto Arabských rodinách; (3)kvůli homozygotnosti nebo složené heterozygotnosti pro mutace byly klinické fenotypy také závažné v našich studovaných rodinách (12-14). Navrhli jsme, že genetická a fenotypová pozorování pramenila z extrémně vysoké míry příbuzenských manželství v Saúdské Arábii (108, 109). Naše studie poskytla první vědecké důkazy o roli pokrevního příbuzenství v vyvíjení klíčovou roli v nevysvětlitelné srdeční arytmie a SCDs u dětí a dospívajících v Saúdské Arábii (12-14). Máme také prokázáno, že mutace KCNQ1 c.387 -5 T > (NM_000218) (Obrázek 3) se rozprostírá v Assir provincii Saúdské Arábie od společného předka během několika generací v důsledku vysoké incidence příbuzenské sňatky . Zatím se jedná o nejčastější kauzální mutaci LQTS1 v saúdskoarabské populaci, která byla také pozorována ve specializované nemocnici krále Faisala a ve vojenské nemocnici Khamis Mashayt (nepublikované). Podobný výsledek byl také získán pro kauzální mutaci LQTS2, c. 3208 C > T (str.Q1070X) (Obrázek 4) v genu KCNH2 (12, 14), který je také zakladatel mutace v Saúdské Arábii a odděleny v průběhu mnoha generací v Assir regionu (viz mapa na Obrázku 6). By jsme předpovídají, že existuje značný počet jedinců s uvedenými předků/zakladatel mutace (obě v KCNQ1 a KCNH2 a jiných genů) v tomto regionu a také ve velkých městech jako je Rijád, Džidda a Dammam, vzhledem k městské migrace. Více zakladatel nebo rodové mutace patogenních pro LQTS jsou velmi plánovaná v jiných provinciích Saúdské Arábie vzhledem k vysoké míře příbuzenské sňatky.
obrázek 6. Mapa Saúdské Arábie (s laskavým svolením: Wikipedia). Oblast Assir je označena tlustou čarou, kde jsme našli zakladatelské mutace v genech kcnq1 a kcnh2, popsaných v rodině 1-4.
Jako LQTS je autosomálně dominantní onemocnění, můžeme pozorovat převážně heterozygotní mutace dopravce pacientů. Ale v naší studii jsme identifikovali hlavně pacienty s recesivními mutacemi (12-14). Je zřejmé, že tito pacienti se stanou prvními symptomatickými a byli převážně odkázáni na Kardiocentrum Prince Sultana, čímž je upozornili. Nicméně, závěry z našeho šetření vyplývá skutečnost, že recesivní LQTS mohlo mít fatální klinických fenotypů u dětí, a oni nejsou neobvyklé v Saúdské Arábii (12-14). Jako heterozygotní mutaci jsou také náchylné k rozvoji arytmie a její komplikace, společné iniciativy by měla být přijata, aby místní obecné lékaři, kardiologové, a klinických genetiků ve společné platformy pro identifikaci jedinců v riziku. Dále v současné době nemáme žádnou genetickou informaci pro jiné poruchy familiární arytmie, např. CPVT, SQTS, BrS, AF atd. Specializovaná kardiogenetická centra by měla převzít iniciativu při hledání genetických defektů, mutací a provádět genotyp-fenotypové studie ve všech formách dědičných arytmií. To také by měly být uchovávány v úvahu, že ne všechny genetické arytmie by mít rodinnou historii, stejně jako v mnoha případech, arytmie kauzální mutace de novo v původu, tj. proband je první pacient, v té rodině s mutací a on/ona je zdrojem k přenosu mutace v navazujících generací (110, 111). Vzhledem k velmi vysoké míře příbuzenské svazky manželství v Saúdské Arábii, očekáváme mnohem víc, zakladatel mutace vyvíjet rozhodující úlohu ve vrozené arytmií v této zemi. Identifikace těchto zakladatelských mutací by měla být naším prvním a nejdůležitějším úkolem, což by nám usnadnilo rozvoj účinného předmanželského a také pre-symptomatického genetického poradenství v této zemi. Mutace v LQTS kauzální geny mohla svěřit variabilita v klinické penetrance, v jednom extrému, někteří dopravci by mohl být údajně úplně zdravé, ale někteří dopravci mohli svůj první projev nemoci jako synkopy nebo náhlé smrti. Náhlá smrt mladé dítě nebo dospělého představuje velkou psychickou a emocionální zátěž na rodinu, promítání dopravci jsou nezbytné, protože existuje jednoduchý lék, např. β-blokátory (také změna chování), což může velmi účinně zabránit dopravci z fatální důsledky arytmie a SCDs. Jedinci s homozygotními mutacemi v kauzálních genech LQTS mohou mít závažnou morbiditu a extrémně vysokou míru úmrtnosti včetně fetálních brady-tachyarytmií a v mnoha případech potratů, spontánních potratů u těhotných matek (12-14).
v Saúdské Arábii nebylo provedeno mnoho výzkumů o prevalenci různých SNP v genech spojených s arytmií a také v genech, které je regulují. Splawski a kol. (112) popsal běžnou variantu S1103Y v genu SCN5A spojenou s arytmií u afroameričanů. Variantní alela označují Y1103 je zodpovědný za urychlení aktivace kanálu, čímž se zvyšuje pravděpodobnost srdeční arytmie u osob Afrického původu (52, 112). K393N je varianta v genu KCNQ1 hlášeny v LQTS1 pacientů v USA, ale v Arabské populace, zjistili jsme, že tato varianta v 2% jedinců (nepublikované údaje). Zda je varianta k393n v genu Kcnq1 u Arabů srovnatelná s variantou S1103y (SCN5A) u afroameričanů nebo variantou D85N (KCNE1) v japonské populaci, by si také mohlo zaslouží vyšetřování (59).
Střet Zájmů Prohlášení
autoři prohlašují, že výzkum byl prováděn v nepřítomnosti jakékoli obchodní nebo finanční vztahy, které by mohlo být chápáno jako potenciální konflikt zájmů.
Poděkování
Naše upřímné poděkování Prof. Arthur Wilde, Prof. Connie Bezzina, a vydavatel časopisu “Srdce” za jejich svolení k použití Obrázku 1 v rukopisu z Ref. (113).
1. Bhuiyan ZA. Klinické a genetické spektrum dědičných syndromů srdeční arytmie. Amsterdam: Amsterdamská Univerzita (2009).
2. Priori SG, Aliot E, Blømstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, et al. Pracovní skupina pro náhlou srdeční smrt, Evropská kardiologická společnost. Europace (2002) 4: 3-18. doi: 10.1053 / eupc.2001.0214
CrossRef Plný Text
3. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, et al. SCN5A mutace spojené s dědičnou srdeční arytmií, syndromem dlouhého QT. Buňka (1995) 80: 805-11. doi:10.1016/0092-8674(95)90359-3
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
4. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. Molekulární základ pro srdeční arytmii: mutace HERG způsobují syndrom dlouhého QT. Buňka (1995) 80: 795-803. doi: 10.1016/0092-8674(95)90358-5
Pubmed Abstraktní | Pubmed Plný Text | CrossRef Celý Text
5. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, et al. Poziční klonování nového genu draselného kanálu: mutace KVLQT1 způsobují srdeční arytmie. Nat Genet (1996) 12: 17-23. doi:10.1038/ng0196-17
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
6. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutace v genu hminK způsobují syndrom dlouhého QT a potlačují funkci IKs. Nat Genet (1997) 17: 338-40. doi:10.1038/ng1197-338
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
7. Sanguinetti MC, Jiang C, Curran ME, Keating MT. Mechanistické spojení mezi zděděnou a získanou srdeční arytmií: HERG kóduje draslíkový kanál IKr. Buňka (1995) 81: 299-307. doi:10.1016/0092-8674(95)90340-2
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. Nová mutace v genu draselného kanálu KVLQT1 způsobuje jervellův a Lange-Nielsenův kardioauditární syndrom. Nat Genet (1997) 15: 186-9. doi:10.1038/ng0297-186
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
9. Kucera JP, Rohr S, Rudy y. lokalizace sodíkových kanálů v interkalovaných discích moduluje srdeční vedení. Circ Res (2002) 91: 1176-82. doi: 10.1161 / 01.RES.0000046237.54156.0
Pubmed Abstraktní | Pubmed Plný Text | CrossRef Celý Text
10. Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar m. Connexin43 remodelace způsobená inhibicí exprese plakophilin-2 v srdečních buňkách. Circ Res (2007) 101: 703-11. doi: 10.1161 / CIRCRESAHA.107.154252
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
11. Rohr s. molekulární přeslechy mezi mechanickými a elektrickými spoji na interkalovaném disku. Circ Res (2007) 101: 637-9. doi: 10.1161 / CIRCRESAHA.107.161901
CrossRef Plný Text
12. Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, et al. Opakující se intrauterinní ztráta plodu v důsledku téměř nepřítomnosti HERG: klinická a funkční charakterizace homozygotní nesmyslné mutace HERG Q1070X. Srdeční Rytmus (2008) 5: 553-61. doi: 10.1016 / j. hrthm.2008.01.020
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
13. Bhuiyan ZA, Momenah TS, Amin TS, Al-Khadra AS, Alders M, Wilde AAM, et al. Intronická mutace vedoucí k neúplnému přeskočení exon-2 v KCNQ1 zachrání sluch u jervella a Lange-Nielsenova syndromu. Prog Biophys Mol Biol (2008) 98: 319-27. doi: 10.1016 / j. pbiomolbio.2008.10.004
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
14. Bhuiyan ZA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi S, Al-Kalaf K, Mannens MMAM a kol. Klinická a genetická analýza syndromu dlouhého QT u dětí ze šesti rodin v Saúdské Arábii: liší se? Pediatr Cardiol (2009) 30: 490-501. doi:10.1007/s00246-008-9377-y
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
15. Shinwari ZM, Al-Hazzani A, Dzimiri N, Tulbah S, Mallawi Y, Al-Fayyadh M, et al. Identifikace nové mutace kcnq1 ve Velké Saúdské rodině se syndromem dlouhého QT: klinické důsledky a preventivní důsledky. Clin Genet (2013) 83: 370-4. doi: 10.1111 / j. 1399-0004. 2012. 01914.x
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence vrozeného syndromu dlouhého QT. Oběh (2009) 120: 1761-7. doi: 10.1161 / CIRCULATIONAHA.109.863209
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
17. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostická kritéria pro syndrom dlouhého QT. Aktualizace. Oběh (1993) 88: 782-4. doi: 10.1161 / 01.Cire.88.2.782
CrossRef Plný Text
18. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. Shrnutí: prohlášení o shodě HRS / EHRA / APHRS o diagnostice a léčbě pacientů s dědičnými syndromy primární arytmie. Europace (2013) 15: 1389-406. doi: 10.1093/europace / eut272
CrossRef Plný Text
19. Liu JF, Jons C, Moss AJ, McNitt S, Peterson DR, Qi M, et al. Registr Syndromu. Rizikové faktory pro rekurentní synkopu a následné fatální nebo téměř fatální příhody u dětí a dospívajících se syndromem dlouhého QT intervalu. J Am Coll Cardiol (2011) 57: 941-50. doi: 10.1016 / j. jacc.2010.10.025
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
20. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spektrum mutací v genech syndromu dlouhého QT. Kvlqt1, HERG, SCN5A, KCNE1 a KCNE2. Oběh (2000) 102: 1178-85. doi: 10.1161 / 01.Cire.102.10.1178
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
21. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Mutace sloučenin: častá příčina těžkého syndromu dlouhého QT. Oběh (2004) 109:1834-41. doi: 10.1161 / 01.Cire.0000125524.34234.13
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
22. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, et al. Riziko život ohrožujících srdečních příhod u pacientů se syndromem dlouhého QT a mnohočetnými mutacemi. Srdeční Rytmus (2013) 10: 378-82. doi: 10.1016 / j. hrthm.2012.11.006
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
23. Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Genetické testování u syndromu dlouhého QT: vývoj a validace efektivního přístupu k genotypizaci v klinické praxi. JAMAOVÁ (2005) 294: 2975-80. doi: 10.1001 / jama.294.23.2975
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
24. Roden DM. Klinická praxe. Syndrom dlouhého QT intervalu. N Engl J Med (2008) 358: 169-76. doi: 10.1056 / NEJMcp0706513
CrossRef Plný Text
25. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotyp-fenotypová korelace u syndromu dlouhého QT: genově specifické spouštěče život ohrožujících arytmií. Oběh (2001) 103: 89-95. doi: 10.1161 / 01.Cire.103.1.89
CrossRef Plný Text
26. Ackerman MJ, Tester DJ, Porter CJ. Plavání, genově specifický arytmogenní spouštěč pro zděděný syndrom dlouhého QT. Mayo Clin Proc (1999) 74:1088-94. doi:10.4065/74.11.1088
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
27. Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, et al. Genetické testování syndromu dlouhého QT: rozlišení patogenních mutací od benigních variant. Oběh (2009) 120: 1752-60. doi: 10.1161 / CIRCULATIONAHA.109.863076
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
28. Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, et al. Klinické aspekty syndromu dlouhého QT typu 1 podle umístění, kódovacího typu a biofyzikální funkce mutací zahrnujících Gen kcnq1. Oběh (2007) 115: 2481-9. doi: 10.1161 / CIRCULATIONAHA.106.665406
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
29. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. Varianty v 3 ‘ nepřeložené oblasti draslíkového kanálu KV7.1 kódovaného kcnq1 modifikují závažnost onemocnění u pacientů se syndromem dlouhého QT typu 1 alelně specifickým způsobem. Eur Heart J (2012) 33: 714-23. doi:10.1093/eurheartj/ehr473
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
30. Barsheshet A, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu W, et al. Mutace v cytoplazmatických smyčkách kanálu KCNQ1 a riziko život ohrožujících příhod: důsledky pro mutační specifickou odpověď na terapii β-blokátory u syndromu dlouhého QT typu 1. Oběh (2012) 125: 1988-96. doi: 10.1161 / CIRCULATIONAHA.111.048041
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
31. Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. Jervellův a Langeův-Nielsenův syndrom: přirozená historie, Molekulární základ a klinický výsledek. Oběh (2006) 113: 783-90. doi: 10.1161 / CIRCULATIONAHA.105.592899
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
32. Bhuiyan ZA, Wilde AA. IKs v srdci a sluchu, ucho může dělat s méně než srdce. Circ Cardiovasc Genet (2013) 6: 141-3. doi: 10.1161 / CIRCGENETICS.113.000143
CrossRef Plný Text
33. Wollnik B, Schroeder BC, Kubisch C, Esperer, Wieacker P, Jentsch TJ. Patofyziologické mechanismy dominantních a recesivních mutací kanálu KVLQT1 k+ nalezené u zděděných srdečních arytmií. Hum Mol Genet (1997) 6: 1943-9. doi: 10.1093/hmg / 6.11.1943
CrossRef Plný Text
34. Wilde AA, Jongbloed RJ, DOEVENDANS PA, Düren DR, Hauer RN, van Langen IM, et al. Sluchové podněty jako spouštěč arytmických příhod odlišují pacienty související s HERG (LQTS2) od pacientů souvisejících s KVLQT1 (LQTS1). J Am Coll Cardiol (1999) 33:327-32. doi:10.1016/S0735-1097(98)00578-6
CrossRef celý Text
35. Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, et al. Zvýšené riziko arytmických příhod u syndromu dlouhého QT s mutacemi v oblasti pórů draslíkového kanálu lidského genu ether-a-go-go. Oběh (2002) 105: 794-9. doi:10.1161/hc0702.105124
Pubmed Abstraktní | Pubmed Plný Text | CrossRef Celý Text
36. Lupoglazoff JM, Denjoy I, Villain E, Fressart V, Simon F, Bozio A, et al. Syndrom dlouhého QT u novorozenců: poruchy vedení spojené s mutacemi HERG a sinusová bradykardie s mutacemi kcnq1. J Am Coll Cardiol (2004) 43: 826-30. doi: 10.1016 / j. jacc.2003.09.049
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
37. Chevalier P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ, et al. Torsades de pointes komplikující atrioventrikulární blok: důkaz genetické predispozice. Srdeční Rytmus (2007) 4: 170-4. doi: 10.1016 / j. hrthm.2006.10.004
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
38. Hoorntje T, Alders M, van Tintelen P, van der Lip K, Sreeram N, van der Wal A, et al. Homozygotní předčasné zkrácení proteinu HERG: knockout lidského HERG. Oběh (1999) 100: 1264-7. doi: 10.1161 / 01.Cire.100.12.1264
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
39. Piippo K, Laitinen P, Swan H, Toivonen L, Viitasalo M, Pasternack M, et al. Homozygotnosti na HERG draslíkových kanálů mutace způsobuje těžkou formou syndromu dlouhého QT: identifikace zjevné zakladatel mutace v Finové. J Am Coll Cardiol (2000) 35: 1919-25. doi:10.1016/S0735-1097(00)00636-7
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
40. Johnson Wh Jr, Yang P, Yang T, Lau YR, Mostella BA, Wolff DJ, et al. Klinická, genetická a biofyzikální charakterizace homozygotní mutace HERG způsobující těžký novorozenecký syndrom dlouhého QT. Pediatr Res (2003) 53: 744-8. diváci: 10.1203 / 01.PDR.0000059750.17002.B6
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
41. Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Etnické rozdíly ve variantách srdečních draslíkových kanálů: důsledky pro genetickou náchylnost k náhlé srdeční smrti a genetické testování vrozeného syndromu dlouhého QT. Mayo Clin Proc (2003) 78: 1479-87. doi:10.4065/78.12.1479
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
42. Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. Kcnh2-K897T je genetický modifikátor latentního vrozeného syndromu dlouhého QT. Oběh (2005) 112:1251-8. doi: 10.1161 / CIRCULATIONAHA.105.549071
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
43. Nof E, Cordeiro JM, Pérez GJ, Scornik FS, Calloe K, Love B, et al. Běžný polymorfismus s jedním nukleotidem může zhoršit syndrom typu 2 s dlouhým QT, což vede k náhlé smrti kojenců. Circ Cardiovasc Genet (2010) 3: 199-206. doi: 10.1161 / CIRCGENETICS.109.898569
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
44. Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, et al. Mutace jednoho kanálu Na (+) způsobující syndrom dlouhého QT i Brugada. Circ Res (1999) 85: 1206-13. doi: 10.1161 / 01.RES.85.12.1206
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
45. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, et al. Nová mutace SCN5A vedoucí buď k izolované defektu srdečního vedení nebo Brugada syndromu ve Velké francouzské rodině. Oběh (2001) 104: 3081-6. doi:10.1161/hc5001.100834
Pubmed Abstraktní | Pubmed Plný Text | CrossRef Celý Text
46. Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, et al. Mutace E1784K u SCN5A je spojena se smíšeným klinickým fenotypem syndromu dlouhého QT typu 3. J Clin Invest (2008)118: 2219-29. doi:10.1172/JCI34057
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
47. Keller DI, Acharfi S, Delacrétaz E, Benammar N, Rotter M, Pfammatter JP, et al. Nová mutace v SCN5A, delQKP 1507-1509, způsobující syndrom dlouhého QT: role zbytku Q1507 při inaktivaci sodíkových kanálů. J Mol Cell Cardiol (2003) 35: 1513-21. doi: 10.1016 / j. yjmcc.2003.08.007
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
48. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Stratifikace rizika u syndromu dlouhého QT. N Engl J Med (2003) 348:1866-74. doi:10.1056/NEJMoa022147
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
49. Lupoglazoff JM, Cheav T, Baroudi G, Berthet M, Denjoy I, Cauchemez B, et al. Homozygotní mutace SCN5A u syndromu dlouhého QT s funkčním atrioventrikulárním blokem dva ku jednomu. Circ Res (2001) 89: E16-21. doi: 10.1161 / hh1401.095087
CrossRef Plný Text
50. Shi R, Zhang Y, Yang C, Huang C, Zhou X, Qiang H, et al. Mutace srdečního sodíkového kanálu delQKP 1507-1509 je spojena s rozšiřujícím se fenotypovým spektrem LQT3, poruchou vedení, dilatační kardiomyopatií a vysokým výskytem náhlé smrti mládeže. Europace (2008) 10: 1329-35. doi:10.1093/europace/eun202
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
51. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartscheer A, Eckardt L, Paul M, et al. Heterozygotní deleční mutace v genu srdečního sodíkového kanálu SCN5A s charakteristikami ztráty a zisku funkce se projevuje jako izolované vodivé onemocnění bez příznaků Brugada nebo syndromu dlouhého QT. PLoS One (2013) 8: e67963. doi: 10.1371 / deník.pone.0067963
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
52. Plant LD, Bowers PN, Liu Q, Morgan T, Zhang T, State MW, et al. Běžná varianta srdečního sodíkového kanálu spojená s náhlou kojeneckou smrtí u afroameričanů, SCN5A S1103Y. J Clin Invest (2006) 116:430-5. doi: 10.1172 / JCI25618
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
53. Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, et al. Mutace Ankyrin-B způsobuje srdeční arytmii typu 4 s dlouhým QT QTEM a náhlou srdeční smrt. Příroda (2003) 421: 634-9. doi:10.1038/nature01335
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
54. Schott JJ, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB, et al. Mapování genu pro syndrom dlouhého QT na chromozom 4q25-27. Am J Hum Genet (1995) 57: 1114-22.
Pubmed Abstrakt / Pubmed Plný Text
55. Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 a lsK (norek) proteiny spojit se tvořit I(Ks) srdeční draslíku aktuální. Příroda (1996) 384: 78-80. doi:10.1038/384078a0
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
56. Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Sestavení K (V)lqt1 a norkových (IsK) proteinů za vzniku srdečního i (Ks) draslíkového kanálu. Příroda (1996) 384: 80-3. doi: 10.1038 / 384080a0
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
57. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al. Mutace kcne1 způsobují jervellův a Lange-Nielsenův syndrom. Nat Genet (1997) 17: 267-8. doi: 10.1038 / ng1197-267
CrossRef Plný Text
58. Duggal P, Veselý MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutace genu pro IKs spojená s jervellovou a Lange-Nielsenovou a Romano-Wardovou formou syndromu dlouhého QT intervalu. Oběh (1998) 97: 142-6. doi: 10.1186/1471-2350-9-24
Pubmed Abstraktní | Pubmed Plný Text | CrossRef Celý Text
59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85n, polymorfismus kcne1, je genová varianta způsobující onemocnění u syndromu dlouhého QT. J Am Coll Cardiol (2009) 54: 812-9. doi: 10.1016 / j. jacc.2009.06.005
PubMed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
60. Paulussen AD, Gilissen RA, Armstrong M, DOEVENDANS PA, Verhasselt P, Smeets HJ, et al. Genetické variace kcnq1, KCNH2, SCN5A, KCNE1 a KCNE2 u pacientů se syndromem dlouhého QT vyvolaným léky. Mol Med (2004) 82: 182-8. doi:10.1007/s00109-003-0522-z
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
61. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 tvoří draslíkové kanály IKr s HERG a je spojena se srdeční arytmií. Buňka (1999) 97:175-87. doi:10.1016/S0092-8674(00)80728-X
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
62. Gordon E, Panaghie G, Deng L, Bee KJ, Roepke TK, Krogh-Madsen T, et al. Mutace kcne2 u pacienta se srdeční arytmií vyvolanou sluchovými podněty a nerovnováhou elektrolytů v séru. Cardiovasc Res (2008) 77:98-106. doi:10.1093/cvr/cvm030
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
63. Omítka NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, et al. Mutace v Kir2. 1 způsobují vývojové a epizodické elektrické fenotypy Andersenova syndromu. Buňka (2001) 105: 511-9. doi: 10.1016/S0092-8674(01)00342-7
Pubmed Abstraktní | Pubmed Plný Text | CrossRef Celý Text
64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primární struktura a funkční exprese myšího usměrňovače draselného kanálu. Příroda (1993) 362:127-33. doi:10.1038/362127a0
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
65. Raab-Graham KF, Radeke CM, Vandenberg CA. Molekulární klonování a exprese lidského srdce dovnitř usměrňovač draselný kanál. Neuroreport (1994) 5: 2501-5. doi: 10.1097/00001756-199412000-00024
Pubmed Abstraktní | Pubmed Plný Text | CrossRef Celý Text
66. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca (V)1.2 dysfunkce kalciového kanálu způsobuje multisystémovou poruchu včetně arytmie a autismu. Buňka (2004) 119:19-31. doi: 10.1016 / j. buňka.2004.09.011
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
67. Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Těžká porucha arytmie způsobená srdečními mutacemi kalciového kanálu typu L. Proc Natl Acad Sci U S A (2005) 102:8089-96. doi: 10.1073 / pnas.0502506102
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
68. Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L, et al. Dlouhé QT, syndaktylie, kontraktury kloubů, mrtvice a nová mutace CACNA1C: rozšíření spektra Timothyho syndromu. Am J Med Genet A (2012) 158A: 182-7. doi: 10.1002 / ajmg.a.34355
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
69. Dufendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. Mateřská mozaika zaměňuje novorozeneckou diagnózu typu 1 Timothy syndrom. Pediatrie (2013) 131: e1991-5. doi: 10.1542 / peds.2012-2941
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
70. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. Mutant caveolin-3 indukuje perzistentní pozdní sodíkový proud a je spojen se syndromem dlouhého QT. Oběh (2006) 114:2104–12. doi: 10.1161 / CIRCULATIONAHA.106.635268
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
71. Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, et al. Román mechanismus pro syndrom náhlého úmrtí kojenců: přetrvávající zpoždění sodíku aktuální sekundární mutace v caveolin-3. Srdeční Rytmus (2007) 4: 161-6. doi: 10.1016 / j. hrthm.2006.11.030
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
72. Engelman JA, Zhang X, Galbiati F, Volonte D, Sotgia F, Pestell RG, et al. Molekulární genetika rodiny genů caveolin: důsledky pro lidské rakoviny, cukrovku, Alzheimerovu chorobu a svalovou dystrofii. Am J Hum Genet (1998) 63: 1578-87. doi:10.1086/302172
CrossRef Plný Text
73. Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Lokalizace srdečních kanálů typu L Ca (2+) do kaveolárního makromolekulárního signalizačního komplexu je nutná pro beta (2)-adrenergní regulaci. Proc Natl Acad Sci U S A (2006) 103: 7500-5. doi: 10.1073 / pnas.0503465103
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
74. Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, et al. Podjednotka beta4 kódovaná sodíkovým kanálem scn4b u vrozeného syndromu dlouhého QT intervalu. Oběh (2007) 116: 134-42. doi: 10.1161 / CIRCULATIONAHA.106.659086
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
75. Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutace proteinu ukotvujícího a-kinázu způsobuje syndrom dlouhého QT intervalu. Proc Natl Acad Sci U S A (2007) 104: 20990-5. doi: 10.1073 / pnas.0710527105
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
76. Wu G, Ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, et al. Mutace alfa-1-syntrofinu a syndrom dlouhého QT intervalu: onemocnění narušení sodíkového kanálu. Circ Arytmus Elektrofyziol (2008) 1:193-201. doi: 10.1161 / oběžník.108.769224
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, et al. Identifikace Kir3.4 mutace vrozeného syndromu dlouhého QT. Am J Hum Genet (2010) 86: 872-80. doi: 10.1016 / j. ajhg.2010.04.017
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
78. Roden DM. Mechanismy a řízení proarytmie. Am J Cardiol (1998) 82:49I–57I. doi:10.1016/S0002-9149(98)00472-X
CrossRef celý Text
79. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. Strukturální základ pro syndrom dlouhého QT vyvolaného léky. Proc Natl Acad Sci U S A (2000) 97: 12329-33. doi: 10.1073 / pnas.210244497
CrossRef Plný Text
80. Kannankeril PJ, Roden DM. Dlouhé QT a torsade de pointes vyvolané drogami: nedávné pokroky. Curr Opin Cardiol (2007) 22: 39-43. doi: 10.1097 / HCO.0b013e32801129eb
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
81. Veerman CC, Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC, et al. Pomalý zpožděný usměrňovač blokáda draselného proudu významně přispívá k syndromu dlouhého QT vyvolaného léky. Circ Arytmus Elektrofyziol (2013) 6: 1002-9. doi: 10.1161 / oběžník.113.000239
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
82. Sesti F, Abbott GW, Wei J, Murray kt, Saksena S, Schwartz PJ, et al. Společný polymorfismus spojený s antibiotiky indukovanou srdeční arytmií. Proc Natl Acad Sci U S A (2000) 97: 10613-8. doi: 10.1073 / pnas.180223197
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
83. Kääb S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, et al. Velký průzkum kandidátských genů identifikuje polymorfismus kcne1 D85N jako možný modulátor torsades de pointes indukovaných léky. Circ Cardiovasc Genet (2012) 5: 91-9. doi: 10.1161 / CIRCGENETICS.111.960930
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
84. Nakamura K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase S, et al. Anti-kcnh2 protilátkami indukovaný syndrom dlouhého QT: nová získaná forma syndromu dlouhého QT. J Am Coll Cardiol (2007) 50: 1808-9. doi: 10.1016 / j. jacc.2007.07.037
CrossRef Plný Text
85. Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, et al. EKG t-vlnové vzory v geneticky odlišných formách dědičného syndromu dlouhého QT. Oběh (1995) 92: 2929-34. doi: 10.1161 / 01.Cire.92.10.2929
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
86. Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, et al. Spektrum vzorců st-T-vln a repolarizačních parametrů u vrozeného syndromu dlouhého QT: nálezy EKG identifikují genotypy. Oběh (2000) 102: 2849-55. doi: 10.1161 / 01.Cire.102.23.2849
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
87. Tan HL, Bardai A, Shimizu W, Moss AJ, Schulze-Bahr E, Noda T, et al. Genotyp specifický nástup arytmií u vrozeného syndromu dlouhého QT: možné důsledky terapie. Oběh (2006) 114: 2096-103. doi: 10.1161 / CIRCULATIONAHA.106.642694
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
88. Locati, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Věk-a sex související rozdíly v klinických projevech u pacientů s vrozeným syndromem dlouhého QT: zjištění z mezinárodního registru LQTS. Oběh (1998) 97: 2237-44. doi: 10.1161 / 01.Cire.97.22.2237
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
89. Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, et al. Vliv genotypu na klinický průběh syndromu dlouhého QT intervalu. Mezinárodní Long-QT syndrom Registry Research Group. N Engl J Med (1998) 339: 960-5. doi: 10.1056/NEJM199810013391404
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
90. Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson DR, McNitt S, et al. Syndrom dlouhého QT po 40 letech. Oběh (2008) 117: 2192-201. doi: 10.1161 / CIRCULATIONAHA.107.729368
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
91. Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, et al. Registr Syndromu. Modulační účinky věku a pohlaví na klinický průběh syndromu dlouhého QT podle genotypu. J Am Coll Cardiol (2003) 42: 103-9. doi:10.1016/S0735-1097(03)00554-0
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
92. Seth R, Moss AJ, McNitt S, Zareba W, Andrews ML, Qi M, et al. Syndrom dlouhého QT a těhotenství. J Am Coll Cardiol (2007) 49: 1092-8. doi: 10.1016 / j. jacc.2006.09.054
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
93. Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, et al. Molekulární souvislost mezi syndromem náhlého úmrtí kojenců a syndromem dlouhého QT. N Engl J Med (2000) 343: 262-7. doi: 10.1056 / NEJM200007273430405
CrossRef Plný Text
94. Schwartz PJ, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, et al. Molekulární diagnostika u dítěte se syndromem náhlého úmrtí kojenců. Lancet (2001) 358: 1342-3. doi:10.1016/S0140-6736(01)06450-9
Pubmed Abstraktní | Pubmed Plný Text | CrossRef celý Text
95. Christiansen M, Tønder N, Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutace v kanálu HERG k+-ion: nová souvislost mezi syndromem dlouhého QT a syndromem náhlého úmrtí kojenců. Am J Cardiol (2005) 95: 433-4. doi: 10.1016 / j. amjcard.2004.09.054
PubMed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
96. Tester DJ, Ackerman MJ. Posmrtný dlouhý Qt syndrom genetické testování náhlé nevysvětlitelné smrti u mladých. J Am Coll Cardiol (2007) 49: 240-6. doi: 10.1016 / j. jacc.2006.10.010
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
97. Hofman N, Tan HL, Clur SA, Alders M, van Langen IM, Wilde AA. Příspěvek zděděného srdečního onemocnění k náhlé srdeční smrti v dětství. Pediatrie (2007) 120: e967-73. doi: 10.1542 / peds.2006-3751
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
98. Wedekind H, Smits JP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, et al. De novo mutace v genu SCN5A spojená s časným nástupem náhlého úmrtí kojenců. Oběh (2001) 104: 1158-64. doi: 10.1161 / hc3501.095361
CrossRef Plný Text
99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, et al. Dysfunkce srdečního sodíkového kanálu u syndromu náhlého úmrtí kojenců. Oběh (2007) 115: 368-76. doi: 10.1161 / CIRCULATIONAHA.106.646513
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
100. Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molekulární a funkční charakterizace nových mutací genu podobného glycerol-3-fosfát dehydrogenáze 1 (GPD1-L) u syndromu náhlého úmrtí kojenců. Oběh (2007) 116: 2253-9. doi: 10.1161 / oběžník.107.704627
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
101. Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, et al. Syndrom náhlého úmrtí kojenců-související mutace v beta podjednotkách sodíkového kanálu. Srdeční Rytmus (2010) 7: 771-8. doi: 10.1016 / j. hrthm.2010.01.032
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
102. Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, et al. Opakující se ztráta plodu ve třetím trimestru a mateřská mozaika pro syndrom dlouhého QT. Oběh (2004) 109:3029-34. doi: 10.1161 / 01.Cire.0000130666.81539.9 E
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
103. Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Ne všechny beta-blokátory jsou stejné v léčbě syndromu dlouhého QT typu 1 a 2: vyšší recidivy událostí pod metoprololem. J Am Coll Cardiol (2012) 60: 2092-6. doi: 10.1016 / j. jacc.2012.07.046
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
104. Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Levá srdeční sympatická denervace při léčbě vysoce rizikových pacientů postižených syndromem dlouhého QT. Oběh (2004) 109:1826-33. doi: 10.1161 / 01.Cire.0000125523.14403.1 E
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
105. Singh B, al Shahwan SA, Habbab MA, al Deeb SM, Biary N. idiopatický syndrom dlouhého QT: kladení správné otázky. Lancet (1993) 341:741. doi:10.1016/0140-6736(93)90501-7
CrossRef celý Text
106. Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. dlouhý QT syndrom s poruchou atrioventrikulárního vedení: maligní varianta u kojenců. Jiří Paroubek (1998) 9: 1225-32. doi: 10.1111 / j. 1540-8167. 1998.tb00096.x
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
107. Kantoch MJ, kurashi MM, Bulbul ZR, Gorgels AP. Novorozenec s komplexním vrozeným srdečním onemocněním, atrioventrikulárním blokem a komorovou tachykardií torsade de pointes. Stimulační Klinický Elektrofyziol (1998) 21: 2664-7. doi: 10.1111 / j. 1540-8159.1998.tb00043.x
CrossRef Plný Text
108. El-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Příbuznost mezi Saúdskoarabským obyvatelstvem. J Med Genet (1995) 32: 623-6. doi: 10.1136 / jmg.32.8.623
CrossRef Plný Text
109. El Mouzan MI, Al Salloum AA, Al Herbish AS, Qurachi MM, Al Omar AA. Příbuznost a hlavní genetické poruchy u saúdských dětí: průřezová studie založená na komunitě. Ann Saudi Med (2008) 28:169-73. doi:10.4103/0256-4947.51726
Pubmed Abstraktní | Pubmed Plný Text | CrossRef Celý Text
110. Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van Tintelen JP, et al. V RYR2-kódovaný ryanodine receptor/calcium release channel u pacientů diagnostikována dříve buď s katecholaminergní polymorfní komorové tachykardie nebo genotyp negativní, cvičení indukované long QT syndrom: komplexní mutační analýza otevřeného čtecího rámce. J Am Coll Cardiol (2009) 54: 2065-74. doi: 10.1016 / j. jacc.2009.08.022
PubMed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
111. Al-Aama JY, Al-Ghamdi S, Bdier AY, Wilde AA, Bhuiyan ZA. De novo mutace v genu KCNQ1 kauzální Jervell a Lange-Nielsen Syndrom. Clin Genet (2013). doi: 10.1111/cge.12300
Pubmed Abstrakt / Pubmed Plný Text / CrossRef Plný Text
112. Splawski I, Timothy KW, Tateyama M, Clancy CE, Malhotra A, Beggs AH, et al. Varianta sodíkového kanálu SCN5A se podílí na riziku srdeční arytmie. Věda (2002) 297: 1333-6. doi: 10.1126 / věda.1073569
CrossRef Plný Text
113. Wilde AA, Bezzina ČR. Genetika srdečních arytmií. Srdce (2005) 91: 1352-8.