Mucopolysaccharidoses: časné diagnostické příznaky u kojenců a dětí
Mucopolysaccharidoses (MPS) jsou skupinou klinicky heterogenní onemocnění způsobené nedostatky lysosomální enzymy potřebné pro odbourávání glykosaminoglykanů (Gag).
MPS jsou charakterizovány progresivními a systémovými klinickými projevy. I přes jejich biochemické a genetické heterogenity, různé druhy sdílet klíčové klinické funkce v různých kombinacích, včetně společných a skeletální dysplazie s tuhostí (kromě MPS IV, kde je laxnost) a bolest, hrubé rysy obličeje, zákalu rohovky, tříselná či břišní kýly, opakující se infekce horních cest dýchacích, onemocnění srdce ventilu, syndrom karpálního tunelu, a variabilní neurologické komplikace. Tyto rysy se obvykle objevují v prvních měsících života u těžkých forem a v raném dětství u oslabených forem, ale často se podceňují a obecně se berou v úvahu pouze tehdy, když jsou jasně patrné.
vzácnosti těchto poruch a variabilita v klinické prezentaci často vede k diagnostickému zpoždění; to může být v rozmezí od měsíců, pro těžké formy, let, pro oslabené (viz Rigoldi et al. v tomto dodatku). Nicméně, vzhledem k rychlé progresi a naléhavou potřebu zásahu v těžké prezentace, a to i několik měsíců zpoždění v diagnostice může mít katastrofální výsledky pro budoucí zdraví pacienta.
Naším cílem je zdůraznit v tomto přehledu velmi časné příznaky závažných forem, které by měly pediatra upozornit na jejich první vzhled.
jaké příznaky / příznaky můžeme očekávat u dětí s těžkými formami MPS?
příznaky a příznaky naznačující různé formy MPS jsou uvedeny v tabulce 1 podle věku. V prvních 6 měsících života, tříselných kýl, abnormálně časté infekce dýchacích cest, zánět středního ucha, a organomegaly může být viděn v MPS pacientů, zejména v MPS I (Hurler syndrom), který má nejvíce předčasná závažné nástup. Navíc, od 6 do 12 měsíců, tyto děti Velmi často vyvinou gibbus (toraco-bederní kyfóza), srdeční šelest, pupeční kýlu, mírnou hypotonii a zpoždění růstu . Mohou mít také typický hrubý vzhled obličeje (obr. 1). MPS II, VI a VII mohou mít podobný časný fenotyp. MPS IV mají pacienti raném kosterní fenotyp s výskytem dysplazie kyčelního kloubu v prvních měsících života a holub hrudník (pectus carinatum) v prvních 12-18 měsíců. Kojenci MPS II jsou přítomni s podobným zapojením, ale s pozdějším nástupem. V druhém roce života, POSLANCI jsem Hurler syndrom děti rozvíjet kognitivní zpoždění, zatímco MPS II pacient může být identifikován pouze kvůli přemnožení, častých infekcí dýchacích cest, a několik operací ; typické rysy obličeje se objeví až později. U některých pacientů s MPS II se také vyvine hyperaktivita mezi 18 a 24 měsíci. Ve třetím roce života, hyperaktivita a kognitivní zpoždění jsou snadno rozpoznatelné v MPS II a MPS III.
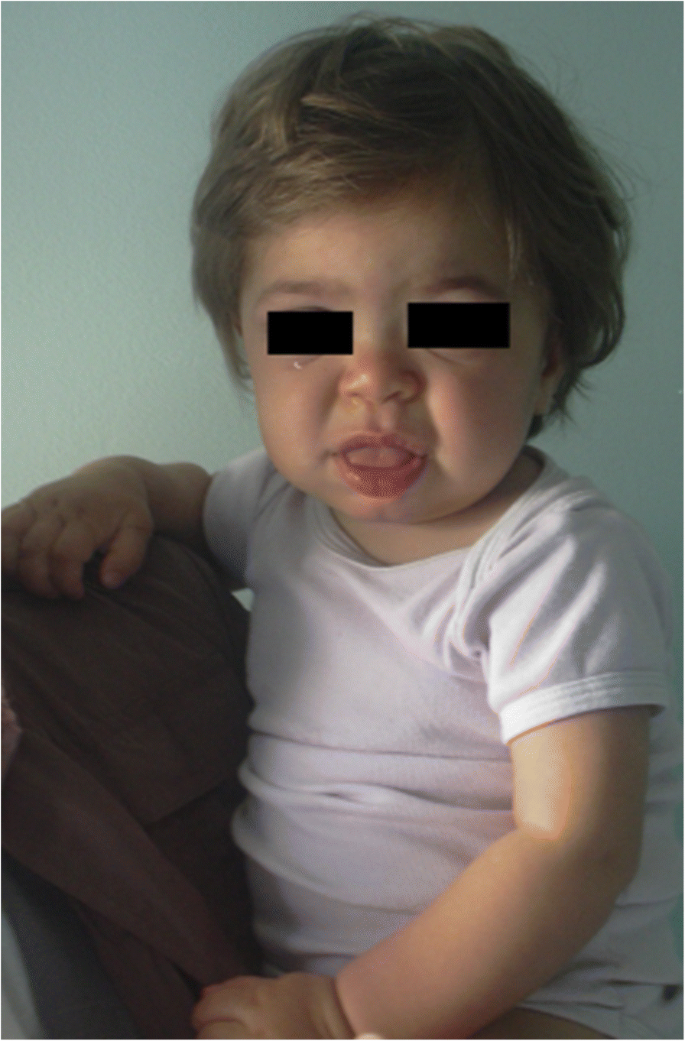
MPS IH u 1letého pacienta. Hrubé facie: plochý nosní můstek, makroglosie, čelní bossing
stručně řečeno, skeletální projevy jsou typické a někdy předčasné. Hrubé facie a kognitivní zpoždění nejsou časnými prominentními znaky.
jaké jsou různé prezentace různých typů poslanců?
POSLANCI se somatické a kognitivní zapojení
Mukopolysacharidóza typu I (MPS I; OMIM #252800) (Obr. 1, 2, 3, 4 a 5) je způsobena deficitem lysozomální hydrolázy, α-l-iduronidázy, který vede k multisystémové hromadění dva Gagy: dermatan sulfátu (DS) a heparan sulfátu (HS) . Na základě své závažnosti je MPS I tradičně rozdělen do tří klinických fenotypů; tyto formy však představují kontinuum závažnosti onemocnění. Hurlerův syndrom, nejzávažnější forma, se obvykle projevuje během prvního roku života. Postižené děti rychle rozvíjejí významné kognitivní poruchy a somatické onemocnění ve více orgánových systémech, což vede k úmrtí během prvního desetiletí bez léčby. Oslabené formy MPS I, známý jako Hurler–Scheie syndrom a Scheie syndrom, respektive, se vyznačují pozdější nástup příznaků, delší průměrná délka života, a mírné nebo žádné centrální nervový systém (CNS) zapojení . Dědičnost je autozomálně recesivní a v nejméně 50% případů je predikce fenotypu možná na základě genotypu, zatímco ve zbývajících 50% jsou detekovány Nové mutace . Prevalence je přibližně 1 ze 100 000 živě narozených .

poslanci IH. Gibbus u 9měsíčního pacienta. b rentgen páteře páteře u 3letého pacienta. C T2-vážený magnetický rezonanční obraz páteře vykazující významnou kyfózu a stenózu páteřního kanálu

poslanci IH. Rentgenový důkaz multiplexu dysostózy. zesílení žeber u 3letého pacienta a b dysplazie kyčle u jiného pacienta ve věku 18 měsíců

MPS IH: Drápová ruka u 18měsíčního pacienta; B ruční rentgen.

poslanci IH. zahušťování kortikální kosti lebky a abnormální konformace sella turcica ve tvaru písmene J. Dilatace periventrikulární prostory v b VKUS a c T-2 vážené magnetické rezonance pacienta ve věku 17 měsíců
Mukopolysacharidóza typ II (MPS II; OMIM #309900) (Obr. 6), také známý jako Hunterův syndrom, je výsledkem nedostatku enzymu iduronát-2-sulfatázy (I2S) s následnou akumulací gag DS a HS . Prevalence je kolem 1 ze 140 000-156 000 živě narozených mužů . Pozoruhodně, toto je pouze X-vázaná POSLANCI, a žena dopravci jsou obvykle asymptomatické; nicméně, oni mohou být velmi ovlivněna v důsledku abnormality na X-chromozomu, homozygotnosti, nebo šikmo-X-inaktivace . Fenotypová exprese také zahrnuje široké spektrum klinické závažnosti. V závažných případech existuje výrazné progresivní neurologické postižení a somatické onemocnění a smrt se obvykle vyskytuje ve druhé dekádě života. Ostatní pacienti s minimální nebo žádné neurologické dysfunkce může představovat závažné somatické zapojení, zatímco na opačném konci spektra, tam jsou pacienti s pouze minimální somatické projevy a normální inteligencí, kteří přežijí do dospělosti . Časné příznaky a symptomy jsou sdíleny těžkými formami MPS I a II.

poslanci II. 3-letý pacient s pouze mírné, charakteristické rysy obličeje
Sporadické případy hydrops fetalis jsou popsány v MPS I , i když, obecně platí, že tyto děti se objeví normální při narození. První známky MPS i se obvykle objevují během prvních měsíců života, zatímco ty v MPS II se obvykle objevují o něco později . Kiely a kol. v retrospektivní studii u 55 Hurlerové pacientů, zjištěno, že respirační příznaky, krmení problémy, tříselná kýla, zánět středního ucha se objevil na střední věk ≤ 6 měsíců, a kyfóza, srdeční abnormality, zvětšení obvodu hlavy, hypotonie, organomegaly, zákalu rohovky, kloubní omezení, a pupeční kýla se objevil mezi 6 a 12 měsíců. Pupeční kýla je jedním z prvních projevujících se rysů u MPS I a II a tříselné kýly jsou hlášeny přibližně u 60% pacientů .
dalším běžným počátečním vodítkem pro MPS I a II mohou být recidivující infekce horních cest dýchacích se zvýšenou sekrecí. Častý otitis, chronická recidivující rýma a přetrvávající výtok z nosu bez zjevné infekce nejsou specifické, ale mohou pomoci při posilování diagnostického podezření, pokud jsou spojeny s konkrétnějšími příznaky. Skladování GAG v oro-hltanu vede ke zvětšení mandlí a adenoidů a přispívá ke komplikacím horních cest dýchacích. Dalším častým nálezem je hlučné dýchání, zejména v noci, Spojené v pozdějších stádiích s obstrukční spánkovou apnoe. V důsledku časté středního ucha a dysostosis z kůstek středního ucha, ztráta sluchu je časté, se obě vodivé a sensorineurální deficity . Tracheobronchomalacie je běžně pozorována a může vést k akutní obstrukci nebo kolapsu dýchacích cest, což je jedna z hlavních příčin úmrtí v následujících letech .
Adenoidektomie, tonzilektomie, a valgózní paty, spolu s kýly jsou častější a včasná chirurgická intervence pro MPS I a II pacientů, často provádí před znát diagnózu (ve více než 50% případů) .
opakující se průjem je poměrně častý v časných stádiích MPS I a může být také přítomen u pacientů MPS II s neurologickým postižením, zatímco u mírně postižených pacientů je neobvyklý .
Gibbusová deformita (dorso-bederní kyfóza) (obr. 2) se klinicky projeví ve středním věku 8 let.6 měsíců až 1 rok v MPS I, což představuje zcela specifický a předčasný ukazatel MPS obecně. Obratlového těla se objeví zploštělé a beaked, což může vést k pozdějším komplikacím, jako jsou míšního nervu zachycení, akutní zranění páteře a atlanto-okcipitální nestability; to znamená, zvláštní péče musí být přijata, aby se zabránilo dislokace atlanto-axiálního kloubu v celkové anestezii a operaci. Po 1 roce věku představují kloubní kontraktury a genu valgum další časté příznaky poškození kloubů a progresivní skeletální dysplazie, které jsou snadno detekovatelné dříve než 2 roky věku u všech kojenců postižených MPS. Všechny tyto kosterní projevy společně, typické pro MPS, se nazývají dysostosis multiplex. Zesílení žeber je vidět na rentgenových snímcích (obr. 3a), dlouhé kosti jsou krátké se širokými hřídeli a kolena jsou náchylná k deformacím valgusu a varusu. Phalangeal dysostosis a synoviální ztluštění vést k charakteristické dráp ruku deformity (Obr. 4), což je další typický rys, který může vyvolat podezření na onemocnění. Dysplazie kyčelního kloubu je často důvodem, proč tyto děti mohou být poprvé viděny ortopedickými specialisty. Typicky, pánev je špatně tvarované, femorální hlavy jsou malé, a contractures valga je běžné, s progresivní a invalidizující deformity kyčle (Obr. 3b).
mezi MPS I a MPS II je pozoruhodný rozdíl; u MPS I se růst kostry zpomaluje o 3 roky věku, zatímco pacienti MPS II vykazují nadměrný růst v prvních 5-6 letech věku a poté zpomalují jejich růst .
Zhrubnutí rysů obličeje (široký nos s rozšířenými nozdrami, výrazné nadočnicové oblouky, velké zaoblené tváře, silné rty, rozšířené vyčnívající jazyk) způsobené ukládání Gag v měkkých tkání orofaciální oblasti a obličejové kosti se projeví během prvních 2 let, medián věku 1.1 let pro Hurler onemocnění a ve věku 18 měsíců a 4 let v závažné formě Hunter onemocnění , i když jemné fenotypové změny mohou být detekovány dříve (již v druhé polovině prvního roku) podle pediatrů s konkrétní zkušeností (Obr. 1). Makrocefalie se stává postupně zřetelnější a scaphocefalie je běžná (obr. 5), ale mikrocefalie je také možná u pacientů s Hurlerem . Hirsutismus obličeje a těla je vždy patrný ve věku 24 měsíců . Kůže může být zesílená a nepružná. Zejména u některých pacientů s MPS II se vyvine výrazná kožní léze, která je popsána jako slonovinové bílé papuly (oblázkové) na horní části zad a po stranách horních paží, patognomonické Hunterova syndromu .
kardiomyopatie a progresivní zahušťování a ztuhnutí chlopňových letáků u pacientů s MPS I mohou vést k mitrální a méně časté aortální regurgitaci . Šelest může být detekován v prvních měsících života a někdy byl hlášen jako první příznak onemocnění . Srdeční postižení je také přítomno téměř u všech pacientů s MPS II, ale začíná to přibližně ve věku 5 let . Onemocnění ventilu může vést k hypertrofii pravé a levé komory a srdečnímu selhání.
výčnělek břicha způsobený progresivní hepatosplenomegalií je snadno patrný po 6 měsících věku, i když skladování gagů v játrech a slezině nevede k dysfunkci orgánů .
zákal rohovky se vyskytuje téměř u všech jedinců s MPS I dříve než 15 měsíců věku, což může způsobit závažné poškození zraku. Naopak zákal rohovky není typickým rysem Hunterova syndromu, ale vyšetření štěrbinovou lampou může odhalit diskrétní léze rohovky, které neovlivňují vidění. Může být detekována dysfunkce sítnice, zatímco glaukom není běžným nálezem.
Brzy psychomotorický vývoj je obecně rozpoznán jako normální, ale v MPS I vývojové zpoždění je obvykle zřejmý do věku 18 měsíců, s progresivní duševní úpadek, což vede k těžkým mentálním postižením. Jazykové dovednosti jsou u těchto dětí velmi omezené. Globální zpoždění vývojových milníků v MPS II se projeví od 2 let . Nicméně, vedoucí neurologické funkce závažné Hunter onemocnění je reprezentován označené poruchy chování, jako je hyperaktivita, tvrdohlavost, agresivita, které jsou obvykle pozorovány u pacientů s oslabenými fenotyp .
Mukopolysacharidóza typ VII (MPS VII; OMIM #253220), také známý jako Sly syndrom, je ultra-vzácné POSLANCI vyznačuje nedostatečnou aktivitou β-glukuronidáza, s lysozomální střádání chondroitin sulfát (CS), DS a HS, což vede k buněčné a orgánové dysfunkce. První pacient byl popsán Sly et al. v roce 1973 bylo v literatuře dosud hlášeno celkem 145 pacientů.
Na klinické prezentaci a progrese onemocnění MPS VII pokrývá široké spektrum závažnosti, od začátku, závažné multisystémové projevy a smrt v prvních měsících, na mírnější fenotyp s pozdějším nástupem, normální nebo téměř normální inteligence, a delší přežití .
Fenotypové charakteristiky pacientů s MPS VII se podobají těm z MPS I a MPS II (malý vzrůst, skeletální dysplazie, makrocefalie, gingivální hypertrofii, opakované ušní infekce, hepatosplenomegalie, kýly, srdeční postižení, snížení plicních funkcí a kognitivních funkcí). Neočekávaně vysoký podíl popsaných pacientů (41%) má v anamnéze neimunní novorozenecké hydrops fetalis (NIHF). Navzdory prenatálnímu nástupu onemocnění u těchto pacientů přežilo 13 z 23 dětství s mírným až středním průběhem do pozdního dospívání. Přítomnost NIHF tedy sama o sobě nepředpovídá možnou závažnost klinického průběhu, pokud pacient přežije dětství. Další časnou známkou je macrocrania, zatímco srdeční chlopně zapojení a opakované infekce horních a dolních cest dýchacích se objevují v prvních 2 letech života. Mírná mentální retardace a progresivní ztráta sluchu s poruchou řeči jsou také patrné s časem.
Stručně řečeno, těžké formy MPS mám velmi časný multisystémový nástup (do 6 měsíců života); MPS II je podobná, ale s pozdějším nástupem, a MPS VII má prenatální nástup s častými NIHF a brzy těžké zapojení horních a dolních cest dýchacích.
POSLANCI se většinou neurologické a kognitivní zapojení
Mucopolysaccharidoses typ III (MPS III, také známý jako Sanfilippo syndrom; Obr. 7) má převládající neurologickou prezentaci. MPS III je porucha lysozomálního skladování způsobená nedostatkem jednoho ze čtyř enzymů zapojených do katabolismu HS. Čtyři různé podtypy jsou známé—MPS III. typu (OMIM #252900), typ B (OMIM #252920), typ C (OMIM #252930), a typ D (OMIM #252940)—každá z důvodu různých enzymů nedostatek: typ A, sulfamidase/SGSH gen; typ B, alfa-N-acetylglucosaminidase/NAGLU gen; typ C, alfa-glucosaminide N-acetyltransferázu/HGSNAT gen; typ D, N-acetilglucosamina-6-solfato sulfatasi/GNS gen). Všechny čtyři typy (a až D) mají autozomálně recesivní dědičnost. MPS III je nejčastější z MPS s odhadovanou prevalencí mezi 0,3 a 4.1 případy pro každých 100 000 novorozenců, v závislosti na podtypu a rase .

poslanci IIIA. Stejný pacient ve věku 2,5 A b 5 let. Poznámka: jinou tvář výraz, ukazuje progresi neurologického poškození
Pacienti s Sanfilippo syndrom show progresivní kognitivní poruchou v druhém a třetím roce života. Na rozdíl od většiny poslanců jsou somatické rysy relativně méně patrné a zapojení CNS je nápadné. Řeč vývoj zpoždění je obvykle první znamení si všiml rodiče, ale hlavní obavy mohou být problémy v chování (hyperaktivita, úzkost a agresivní chování, poruchy spánku), následuje progresivní duševní úpadek . Tyto příznaky mohou způsobit chybné diagnóze, jako poruchy chování, poruchy pozornosti s hyperaktivitou (ADHD) a poruchami autistického spektra . Může být také přítomna epilepsie.
poruchy spánku, jejichž výskyt je hlášen až 80-90%, jsou běžným rysem. Skládají se z obtíží při usínání a častého nočního probuzení, s úplným obrácením denního a nočního rytmu u některých pacientů.
všechny tyto projevy jsou velmi obtížné zvládnout a mají obrovský psychologický dopad na kvalitu života celé rodiny.
obecně, i když fenotyp může být velmi podobné ve čtyři podtypy, klinický průběh v Sanfilippo B a C se zdá být charakterizován méně závažným fenotypem .
non-neurologické příznaky jsou obvykle méně výrazné u MPS III než u ostatních MPS. Opakující se infekce uší, nosu a krku jsou pozorovány v mladším věku. Opakující se otitis, defekty středního ucha a abnormality ve vnitřním uchu mohou způsobit hluchotu. Přebytek hustých sekrecí a anatomických změn může způsobit obstrukci dýchacích cest. V prvních letech života je často hlášen průjem, zatímco zácpa je častější u starších pacientů.
fyzikální vyšetření může vykazovat hrubé rysy obličeje, i když jsou méně výrazné. Pacienti mají makrocefalie, široké obočí s mediální spalování a synophrys, vlasy jsou většinou suché a hrubé, a hypertrichóza je běžné. U mladších pacientů jsou často pozorovány mírné hepatomegalie a pupeční a inguinální kýly. Vzácné a často mírné kontraktury se většinou vyskytují v loktech. Růst je ovlivněn přibližně u poloviny pacientů starších 12 let .
stručně řečeno, MPS III nebo Sanfilippo nemoc má prominentní neurologické postižení s velmi mírnými somatickými příznaky. Batolata ve věku 2-3 let staré rozvojových hyperaktivita, ADHD, a agresivní chování může být podezření, že POSLANCI III.
POSLANCI s převládající somatické zapojení
MPS MPS IV a VI přítomna pouze somatické zapojení. Jedná se o progresivní podmínky, které šetří intelektuální schopnosti a ovlivňují hlavně kostru (MPS IVA)nebo všechny orgány / systémy těla (MPS VI). Dědičnost je autozomálně recesivní. Míra zhoršení příznaků se u postižených jedinců liší.
mukopolysacharidóza IV (MPS IV, také známá jako Morquio syndrom; obr. 8) uvádí jako dvě geneticky odlišné poruchy, každý s různých enzymů nedostatek: MPS IVA (Morquio syndrom; OMIM #253000) vzhledem k nedostatku N-acetylgalaktosamin-6-sulfatázy (GALNS gen), což vede k hromadění keratan-sulfátu (KS) a CS ; a MPS IVB (Morquio B syndrom; OMIM #253010) vzhledem k nedostatku beta-galaktosidázy což vede k moči VM a oligosacharid vylučování jako v GM1 pacientů. MPS IVB je skutečně alelická k různým formám GM1-gangliosidózy, ale postrádá psychomotorické zhoršení pozorované u GM1 gangliosidózy .

poslankyně IVA. 4 letého pacienta s b antero-posteriorní a c boční x-ray ukazuje “pádlo ve tvaru” žebra a zploštělé a zaoblené obratlů s typickým “přední beaking” aspekt
u Pacientů s těžkou formou MPS IVA s žádnou detekovatelnou aktivitu enzymu odhalit jejich první příznaky se obvykle během prvního roku života, ačkoli oni jsou zřídka diagnostikována před 4-5 roky věku. Informace o přirozené anamnéze pacientů s Morquiem A jsou odvozeny hlavně ze dvou rozsáhlých sbírek údajů . Snížená rychlost růstu začíná přibližně ve věku 18 měsíců a růst se zastaví přibližně ve věku 7 nebo 8 let. Na rozdíl od ostatních MPS jsou klouby Morquio A obvykle laxní a velmi flexibilní (hypermobilní), ale některé klouby (obvykle boky a ramena) mohou mít omezený rozsah pohybu. U těchto pacientů kosterní zapojení obdržet diagnózu, nebo podstoupit hodnocení, spondyloepiphyseal dysplazie, pseudoachondroplasia, více epifyzeální dysplazie, nebo bilaterální Legg-Calvé-Perthes .
konstantním znakem je odontoidní hypoplazie; tak, dislokace atlanto-okcipitálního kloubu může dojít, což způsobuje komprese a poškození bulbární a míchy, což vede k ochrnutí nebo dokonce smrt, pokud krční stabilizace není provedeno brzy.
pacienti s MPS IVB mají velmi podobný klinický projev, ale s méně prominentním krátkým postavením. Mají mírně hrubé rysy obličeje, ztrátu sluchu, zákal rohovky, chlopňové srdeční onemocnění a časté infekce horních cest dýchacích. Mohou se také projevit inguinální kýlou a mírnou hepatomegalií. Kosterní dysplastické rysy patří platyspondylie, odontoid hypoplazie s možným subluxace krční, kypho-skolióza, contractures valga, a přiškrcený kyčelní křídla .
mukopolysacharidóza VI (MPS VI, také známý jako Maroteaux-Lamy syndrom; OMIM #253200; obr. 9) je způsobena patogenními mutacemi v genu arylsulfatázy B (ARSB) umístěném na chromozomu 5. V důsledku toho, snížené nebo chybějící aktivitou enzymu arylsulfatáza B (ASB) (N-acetylgalaktosamin-4-sulfatázy) zhoršuje degradaci DS stanovení jeho buněčné akumulace .

MPS VI. 2-rok-starý chlapec s evidentní hrubý obličej a kosterní dysostosis
Pacienty bez detekovatelné aktivity enzymu mají těžkou formou MPS VI a vykazují příznaky v raném dětství. Jejich somatický fenotyp je podobný hurlerovu syndromu. Ve většině případů se ve věku 2 nebo 3 let projeví závažné a progresivní poškození skeletu, nazvané dysostosis multiplex. Tento kosterní dysplazie patří dysplazie kyčelního kloubu s dysplastických stehenní kosti, abnormální obratlů, klíční kosti nepravidelné, hypoplastické distální vřetenní a loketní kosti a dysplastických a krátké záprstní kosti; karpální a tarsální kosti jsou hypoplastické a mají nepravidelný profil. Rychlost růstu se často zpomaluje po prvním roce života, přičemž konečná výška je obecně nižší než 120 cm. Stejně jako u ostatních poslanců je časným znakem hrubé rysy obličeje. Kromě toho mají pacienti typické vyčnívající břicho, hepatomegalii, pupeční a/nebo inguinální kýlu a postižení srdce v raném dětství. Ačkoli není přítomno žádné primární mentální postižení, u některých pacientů s následnou intrakraniální hypertenzí a papilémem lze pozorovat hydrocefalus. Pectus carinatum, tuhé a stahované klouby, skolióza a kyfóza a komprese krční míchy se časem zhoršují .
stručně řečeno, poslanci IV a VI nevykazují zapojení CNS. MPS IV je jedinečný MPS ve vykazování společné laxnosti, zatímco závažný MPS VI nástup je podobný tomu, který byl pozorován u MPS i.