artikel
Josivan Gomes Lima1*, Marcel Cat Larro Ferreira dos Santos1, Julliane Tamara Ara Larro de Melo Campos2
1departamento de medicina Cl prisnica, disciplina de endocrinologia e metabologia. Hospital universitet Kristirio Onofre Lopes, Universidade Federal do Rio Grande do Norte (UFRN), Natal, Rn, Brasilien
2fakultetet for Sundhedsvidenskab i Trairi, Federal University of Rio Grande do North (UFRN), Natal, Rn, Brasilien
abstrakt
medfødt generaliseret lipodystrofi (CGL) er en sjælden og alvorlig autosomal recessiv sygdom. Patienter er defekte i opbevaring af kropsfedt, og derfor deponerer de fedt i ektopiske væv, hovedsageligt lever, og kan udvikle skrumpelever. Insulinresistens er et typisk fund, forårsager diabetes, der kræver høje daglige doser insulin. I staten Rio Grande do Norte, Brasilien, har vi en af de største kohorter af patienter med CGL. I denne artikel gennemgår vi patofysiologi, klinisk billede og behandling af denne sygdom.
introduktion
type 2-diabetes er et verdenssundhedsproblem og skyldes normalt overdreven vægt og øget visceralt fedt, der forårsager perifer insulinresistens og en manglende evne til bugspytkirtlen til at frigive insulin for at kompensere denne resistens. Andre mindre almindelige typer diabetes forekommer på grund af specifikke genetiske mutationer, som den medfødte generaliserede lipodystrofi (CGL), også kendt som Berardinelli-Seip medfødt lipodystrofi (BSCL). CGL er en autosomal recessiv sygdom, der er klassificeret i fire typer, baseret på genmutation. De ændrede gener spiller vigtige funktioner til dannelse af adipocytter, lipidproduktion og korrekt opbevaring inde i adipocyten. Mutationerne mindsker fedtvæv med deraf følgende aflejring af fedt på ektopiske steder, hvilket forårsager fedtlever, ændret kulhydratmetabolisme, svær insulinresistens med hyperinsulinæmi og akromegaloidegenskaber og dyslipidæmi1-3. CGL-syndromet har omkring 500 tilfælde rapporteret i verden. I Brasilien, i staten Rio Grande do Norte (RN), har vi diagnosticeret, behandlet og fulgt 54 tilfælde i de sidste 20 år4, 5. I en beskrivende undersøgelse ved hjælp af sekundære data estimerede vi i alt 103 patienter i RN6. Dette indikerer en meget højere prævalens end den, der er rapporteret i litteraturen (1: 1 million) 7.
dannelse og opbevaring af Triacylglycerol i lipiddråber
biosyntesen af triglycerider og phospholipider (figur 1a) starter med glycerol-3-phosphatacyltransferase (GPAT), der acylerer glycerol-3-phosphatet i position 1 og danner 1-Acylglycerol-3-phosphat (lysophosphatidsyre). Det efterfølges af et andet acyleringstrin i position to af AGPAT (1-Acylglycerol-3-phosphatacyltransferase) med oprindelse i 1,2-Diacylglycerol-3-phosphat (phosphatidsyre). Det er et vigtigt mellemliggende trin i biosyntesevejen for både triglycerider og phosphoglycerider. Der er 11 isoformer af AGPAT, kodet af forskellige gener4. AGPAT1 og AGPAT2 er de mest omfattende undersøgte. AGPAT1 er til stede i høje niveauer i testis, bugspytkirtel og i mindre grad i fedtvæv og andre væv som hjerte, placenta, hjerne, lunge, mens AGPAT2 er rigeligt i fedtvæv. I de følgende trin stammer den cytosoliske phosphatidsyrefosfatase (PAP eller lipin) fra 1,2-diacylglycerol, og 1,2-diacylglycerolacyltransferase (dgat) danner triacylglycerol4. Phosphatidinsyre og diacylglycerol kan også stamme fra andre phospholipider, såsom cardiolipin, phosphatidylinositol og phosphatidylcholin.
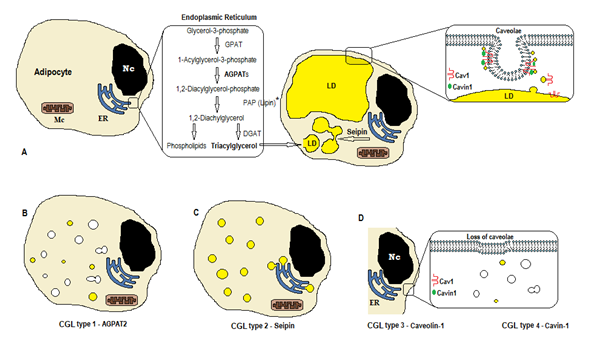
Figur 1. Skema af triglyceridsyntese ifølge CGL-typer. A) normal syntese og opbevaring af triacylglycerol (TAG) i adipocytten. (B) Mutation af AGPAT2 nedsætter TAGPRODUKTIONEN (nogle syntetiseres stadig under stimulering af andre AGPATs). (C) Mutation af seipin gen fald TAG syntese og lipid dråbe (ld) dannelse og fusion. D) Caveolin – 1 og Cavin-1 er nødvendige for dannelsen og stabiliseringen af caveolae. Mutation i CAV1 (type 3) eller CAVIN1 (type 4) kan forårsage tab af caveolae i membranen. Nc, nucleus. ER, endoplasmatisk retikulum. Mc, mitokondrier. * Lipin er et cytosol, der er forankret af seipin i ER.
disse reaktioner forekommer i adipocytternes endoplasmatiske retikulum (ER), hvor en progressiv ophobning af triglycerider forårsager dannelsen af små lipiddråber (LD)8. Produktet af genet BSCL2 er et transmembranprotein kaldet seipin, der forårsager fusion af lille LD med oprindelse i stor ld. Seipin ligger i ER og koncentrerer sig ved krydset med spirende LD, hvilket letter lipidtrafikken mellem ER og LD og inkorporering af triglycerider i LD9. Seipin kan også fungere som et er-anker til det cytosoliske Lipin 1. Udover at være nødvendigt for lipiddråbefusion, størrelse og morfologi er seipin også afgørende for adipogenese (via interaktion med lipin 1) og cellulær triglycerid lipolyse10, 11. Mangel på seipin hæmmer differentieringen af præ-adipocytter til adipocytter og påvirker den endelige modning9, som vist ved undersøgelser i mesenkymale stamceller med bscl2 slået ud12. Ikke-fedtvæv udtrykker også seipin, og andre funktioner skal bestemmes.
i adipocytterne udgør caveolae, som er specialiserede 50-100 nm membraninvaginationer, 20% af plasmamembranområdet, hvilket gør adipocytterne til cellerne med den højeste tæthed af caveolae13. Dannelsen af lipiddråber har brug for et membranprotein (Caveolin – hovedkomponenten i caveolae-membraner) og et cytoplasmatisk protein (Cavin-1)14. Generne CAV1, CAV2 og CAV3 koder for tre former for caveolin med lignende strukturer (henholdsvis Caveolin-1, Caveolin-2 og Caveolin-3). Caveolin – 1 og Caveolin-2 er til stede i adipocytter, fibroblast og endotelceller, og Caveolin-3 er kun til stede i skelet-og hjertemuskler13, 15. Caveolin-1 er den vigtigste og mest studerede. Det udtrykkes i to forskellige isoformer (1A og 1b). Caveolin – 1 translokeres fra plasmamembranen til lipiddråben, hvilket er nødvendigt for lipidhandel og metabolisme16. Lipiddråber opbevarer triglycerider efter fodring, og disse molekyler hydrolyseres til fedtsyre og frigives under faste; denne mekanisme kan reguleres af Caveolin-116. Caveolin – 1-mangel øger også følsomheden over for celledød ved autofagi17.
genet CAVIN1 koder for et cytoplasmatisk protein kaldet caveolae-associeret protein 1 (Cavin-1)14, 16, Der er obligatorisk til dannelse og stabilisering af caveolae. Cavin – 1 udtrykkes i adipocytter, muskelceller og andre celler, og er også afgørende i transmissionen af caveolae-stammer signaler14, 18. Knockout af CAV1-genet forårsager mangel på caveolae i ikke-muskelceller, mens knockout af CAVIN1 forårsager fraværet af caveolae i alle væv, herunder muskler14. Manglen på caveolae kan påvirke reguleringen af lipolyse, fedtsyrestrøm, triglyceridsyntese og signalerne fra andre veje.
typer af CGL
baseret på påviselige genetiske ændringer beskrives fire typer. Type 1 og 2 er ansvarlige for over 95% af tilfældene, og type 2 har en mere alvorligt påvirket fænotype. Kun et tilfælde af type 3 og omkring 30 tilfælde af type 4 er blevet rapporteret4.
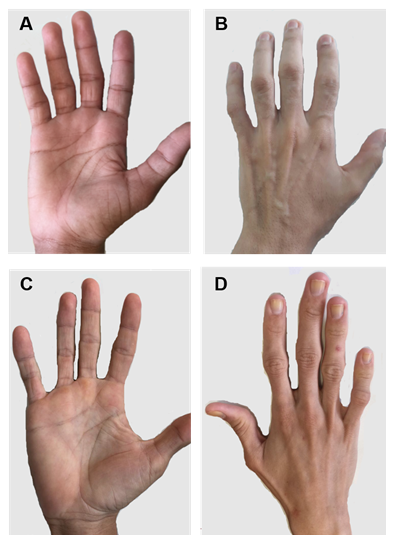
figur 2. Patienter med CGL type 1 og 2. (A) og (B) anterior og posterior udsigt over hænderne på type 1-patienter. Tilsyneladende normale hænder, da der stadig er mekanisk fedtvæv. (C) og (D) anterior og posterior udsigt over hænderne på type 2 patienter. Sværhedsgraden af sygdommen er større, og manglen på fedt er tydelig og let mærkbar.
CGL Type 1. I 1999, Garg et al. 9K34, og tre år senere Agarval et al. viste AGPAT2 som det påvirkede af denne mutation2, 19. På grund af mutation af denne AGPAT2 sker ingen eller minimal produktion af triacylglycerol ved stimulering af andre isoformer. Fænotypen af agpat2-knockout-mus svarer til den hos mennesker med CGL-type, hvilket bekræfter denne fsymes rolle i patofysiologien20, 21.
CGL Type 2. Magre et al. var de første til at identificere mutationen i seipin-genet (kromosom 11k13)3. Mutationer (for det meste nonsens) af seipingenet (BSCL2) producerer et trunkeret protein og kan påvirke lipidmetabolisme ved forskellige mekanismer: a) fald i seipinstabilitet; b) reduktion i evnen til at binde lipin 1; og c) manglende oligomerisering og lokalisering udelukkende til er-membranen11. Nogle celler er stadig i stand til at generere triacylglycerol og små lipiddråber, men store lipiddråber er fraværende på grund af tab af evnen til fusion af disse små lipiddråber. Der er også en fejl i ekspressionen af adipogene faktorer, såsom peroksisom proliferator-aktiveret receptor gamma (PPARG), såvel som adiponectin og adipocyt fedtsyrebindende protein (FABP4)11, 16. Seipinmangel forringer adipogenese, øger lipolyse og forhindrer akkumulering af triglycerider i adipocytter.
CGL type 3. Denne type blev for nylig beskrevet hos en patient, der på trods af at have CGL-fænotype ikke havde mutationer i gener AGPAT2 eller BSCL222. Mus med en mutation i Cav1 er resistente over for diætinduceret fedme og har insulinresistens, hypertriglyceridæmi, nedsat adiponectin, reduceret fedtmasse og små adipocytter16. Efter at have valgt kandidatgener baseret på studier på mus, Kim et al. bekræftet tilstedeværelsen af nonsens mutation i caveolin-1 genet (CAV1) på kromosom 7k3122.
CGL type 4. I dette er en sjælden type det berørte gen er CAVIN1, som koder for proteinet Cavin-1. Hos mennesker er det rapporteret hos patienter med generaliseret medfødt lipodystrofi og muskeldystrofi15, 23.
for nylig er mutationer i PCYT1A-og PPARG-generne også blevet beskrevet, hvilket forårsager lipodystrophy24, 25.
kliniske træk
CGL-patienter præsenterer normalt akromegaloid facies, acanthosis nigricans, phebomegali, hepatomegali og muskelhypertrofi5, 26, 27. Flere forfattere nævner navlebrok som et klinisk fund af syndromet26. Vi vurderede hyppigheden af det i vores serie af patienter, og ingen af dem præsenterede denne ændring28. Faktisk forårsager fraværet af periumbilisk fedtvæv fremspring af navlestrengen, og dette kan fejlagtigt diagnosticeres som en brok28, 29.
når adipocytter ikke kan lagre fedt tilstrækkeligt, akkumuleres det i andre væv som lever og muskler, hvilket forårsager alvorlig insulinresistens. Knogledensitometri kan vise normal eller høj knoglemineraltæthed30 og reduceret total kropsfedt (normalt lavere end 6%)27. Som følge af lavt kropsfedt er serum adiponectin og leptin også lave27. Da leptin er afgørende for at kontrollere sult, har disse patienter typisk hyperfagi, hvilket er tydeligt siden barndommen. Adiponectin spiller en vigtig rolle som en insulinsensibilisator, og dens mangel forværrer insulinresistensen. På trods af dette er glucose og glyceret hæmoglobin i første omgang normalt på bekostning af meget høje insulinniveauer. Diabetes starter normalt i puberteten; i vores serie var gennemsnitsalderen for begyndelsen 15,8 kr. 7,1 år27. Oprindeligt kontrolleres de med orale lægemidler, der har brug for høje doser insulin om få år27. Arteriel hypertension forekommer hos en tredjedel af patienterne27.
der er nogle specifikke kliniske træk ved hver CGL-type. Patienter med type 1 har stadig mekanisk fedtfedt, især i palmer, såler, orbital, peri-artikulære regioner31. I modsætning hertil viser type 2-patienter et fravær af metabolisk og mekanisk fedtvæv. Seipin er stærkt udtrykt i hjernen og cerebellum og er også involveret i reguleringen af neurale funktioner. Mere end halvdelen af type 2-patienter har en vis kognitiv svækkelse1, 8. Type 3 og 4 har konservering af mekanisk og knoglemarvsfedt, og type 4 har muskelsvaghed forbundet med høj serumkreatinkinase og spinal ustabilitet15.
der er også kønsspecifikke kliniske træk. Polycystiske ovarier og amenorrhea er almindelige32. Menstruationscyklusser vender normalt tilbage til normal ved brug af metreleptin, sandsynligvis på grund af forbedring af insulinfølsomhed og restaurering af LH-pulsatility32. Type 2 mænd kan have teratosoospermi på grund af manglen på seipin i kimceller33.
hypertriglyceridæmi forekommer siden de første leveår og kan forårsage akut pancreatitis. HDL er normalt lavere end 30 mg/dL. Forhøjelser af lever er også et tidligt fund og kommer fra fedtaflejringen i leveren. Progressive reduktioner i serumpladerne tyder på forværring af leversygdommen og sandsynlig cirrose34.
da Cavin-1 er til stede i muskelcellerne, har patienter med type 4 mild muskelsvaghed og forhøjet kreatinkinase15.
forventet levealder, hovedsageligt i type 2, er væsentligt nedsat, idet døden ikke sjældent forekommer før 30 år (personlig erfaring baseret på 20 patienter, der døde i de sidste 19 år). Dødsårsagerne er relateret til diabetes (nyresvigt, pludselig død), lever (skrumpelever, fordøjelsesblødning) eller infektioner.
diagnose og behandling
CGL-diagnosen er baseret på kliniske data: akromegaloidegenskaber, acanthosis nigricans, reduktion af total kropsfedt, muskelhypertrofi og fremspring af navlestrengen. Laboratoriedata kan også vise diabetes med svær insulinresistens og hypertriglyceridæmi. Billeddannelsestest kan hjælpe med at identificere ektopiske aflejringer af fedt hovedsageligt i leveren og bugspytkirtlen (leversteatose med hepatomegali og bugspytkirtelsteatose). DKSA kan bekræfte lavt kropsfedt og høj knogletæthed30.
behandlingen af CGL består af streng kontrol af kosten med faldet i indtagelsen af fedt, hovedsageligt triglycerider og fødevarer med et højt glykæmisk indeks for at forebygge og kontrollere comorbiditeter29. Den ideelle diæt er imidlertid et udfordrende mål at opnå på grund af den øgede appetit og den alvorlige begrænsning, der anbefales. Fysisk aktivitet bør også tilskyndes til at forbedre kontrollen med comorbiditeter, undtagen hos patienter med kontraindikationer såsom svær kardiomyopati29.
med hensyn til lægemiddelbehandling kan disse patienter behandles med de sædvanlige medicin mod diabetes, hypertension og dyslipidæmi retningslinjer. Det første valg til behandling af diabetes og insulinresistens er metformin, men normalt er det ikke nok. I modsætning til behandling af partiel lipodystrofi bør thiasolidinedioner anvendes med forsigtighed29. Andre orale antidiabetika anvendes, men de blev ikke specifikt undersøgt hos CGL-patienter. Der er data hos dyr, der tyder på, at brugen af SGLT2-hæmmere kan have fordele, der forhindrer kardiomyopati35; undersøgelser er nødvendige for at bekræfte dette hos mennesker. Efterhånden som sygdommen skrider frem og alvorlig insulinresistens opstår, er der behov for høje daglige doser insulin. Manglen på subkutant fedtvæv er et problem ved indgivelse af de høje doser insulin. Mere koncentreret insulin (U-300 eller U500) kan være påkrævet36. Disse patienter har svær dyslipidæmi, hovedsageligt på grund af stigningen i triglycerider og lav HDL, og derfor er brugen af fibrat undertiden nødvendig for at forhindre akut pancreatitis. På grund af den høje kardiovaskulære risiko for disse patienter bør intervention med et statin overvejes, og målene for LDL eller ikke-HDL bør være strenge29.
daglige injektioner af metreleptin forårsager et signifikant fald i appetitten og giver fordele ved at sænke glykæmi, triglyceridæmi og lever. Det er bemærkelsesværdigt, især hos børn, reduktionen af abdominal omkreds, sandsynligvis på grund af en reduktion af hepatomegali.
konklusion
CGL er en sjælden og alvorlig sygdom, der kan forekomme med diabetes (normalt kræver høje doser insulin) og tidlig død. Patientens fænotype er ret karakteristisk, hvilket dog kræver kendskab til syndromet af sundhedspersonalet for at stille en tidlig diagnose. Metreleptin synes at være den eneste medicin i øjeblikket, der kan ændre sygdommens naturlige historie.
interessekonflikt: ingen.
- Nolis T. udforskning af patofysiologien bag de mere almindelige genetiske og erhvervede lipodystrofier. Tidsskrift for human genetik. 2014 Jan; 59 (1): 16-23.
- Agarval AK, Arioglu E, de Almeida S, et al. AGPAT2 er muteret i medfødt generaliseret lipodystrofi forbundet med kromosom 9K34. Nat Genet. 2002 maj; 31 (1): 21-3.
- Magre J, Delepine M, Khallouf E, et al. Identifikation af genet ændret i Berardinelli-Seip medfødt lipodystrofi på kromosom 11k13. Naturgenetik. 2001 August; 28 (4): 365-70.
- Patni N, Garg A. Medfødte generaliserede lipodystrofier-ny indsigt i metabolisk dysfunktion. Nature anmeldelser endokrinologi. 2015 September; 11 (9): 522-34.
- Garg A. erhvervede og nedarvede lipodystrofier. Det Nye England journal of medicine. 2004 Mar 18; 350(12): 1220-34.
- De Medeiros lb, Candido Dantas VK, Craveiro Sarmento AS, et al. Høj forekomst af Berardinelli-Seip medfødt lipodystrofi i staten Rio Grande do Norte, nordøst Brasilien. Diabetol Metab Syndr. 2017; 9: 80.
- Chikette E, Oral EA, Garg A, et al. Estimering af forekomsten af generaliseret og delvis lipodystrofi: fund og udfordringer. Diabetes, metabolisk syndrom og fedme: mål og terapi. 2017: 375-83.
- lille K, Yang V, Sugii s, et al. Mod en mekanistisk forståelse af lipodystrofi og seipin funktioner. Bioscience rapporter. 2014; 34(5).
- dullet L, Magre J, Cariou B, et al. Funktion af seipin: ny indsigt fra Bscl2 / seipin knockout musemodeller. Biochimie. 2014 Jan; 96: 166-72.
- Sim MF, Dennis RJ, Aubry EM, et al. Det humane lipodystrofiprotein seipin er en er-membranadapter til den adipogene pa-phosphatase lipin 1. Molekylær metabolisme. 2012; 2(1): 38-46.
- Sim MF, Talukder MM, Dennis RJ, et al. Analyse af naturligt forekommende mutationer i det humane lipodystrofiprotein seipin afslører flere potentielle patogene mekanismer. Diabetologia. 2013 November; 56 (11): 2498-506.
- Payne VA, Grimsey N, Tuthill A, et al. Det humane lipodystrofigen BSCL2 / seipin kan være essentielt for normal adipocytdifferentiering. Diabetes. 2008 August; 57 (8): 2055-60.
- Cohen av, Hnasko R, Schubert et al. Rolle caveolae og caveolins i sundhed og sygdom. Fysiologiske anmeldelser. 2004 oktober; 84 (4): 1341-79.
- Pilch PF, Liu L. Fat caves: caveolae, lipidhandel og lipidmetabolisme i adipocytter. Tendenser inden for endokrinologi og metabolisme: TEM. 2011 August; 22(8): 318-24.
- Hayashi YK, Matsuda C, Oga M, et al. Humane ptrf-mutationer forårsager sekundær mangel på caveoliner, hvilket resulterer i muskeldystrofi med generaliseret lipodystrofi. J Clin Invest. 2009 Sep; 119 (9): 2623-33.
- Parton RG, del Poso MA. Caveolae som plasmamembran sensorer, beskyttere og arrangører. Naturen gennemgår Molekylær cellebiologi. 2013 Februar; 14 (2): 98-112.
- Le Lay S, Briand N, Blouin CM, et al. Den lipoatrofiske caveolin-1-mangelfulde musemodel afslører autofagi i modne adipocytter. Autofagi. 2010 August; 6 (6): 754-63.
- Liu L, brun D, McKee M, et al. Sletning af Cavin / PTRF forårsager globalt tab af caveolae, dyslipidæmi og glukoseintolerance. Cellemetabolisme. 2008 oktober; 8 (4): 310-7.
- Garg A, Barnes R, et al. Et gen for medfødt generaliseret lipodystrofi kortlægges til humant kromosom 9K34. Journal of clinical endocrinology and metabolism. 1999 Sep; 84 (9): 3390-4.
- Vogel P, Læs R, Hansen G, et al. Patologi af medfødt generaliseret lipodystrofi hos Agpat2 – / – mus. Veterinær patologi. 2011 kan; 48 (3): 642-54.
- Cortes VA, Curtis de, Sukumaran S, et al. Molekylære mekanismer for hepatisk steatose og insulinresistens i agpat2-mangelfuld musemodel af medfødt generaliseret lipodystrofi. Cellemetabolisme. 2009 februar; 9 (2): 165-76.
- Kim CA, Delepine M, Boutet E, et al. Forening af en homosygøs nonsens caveolin – 1-mutation med Berardinelli-Seip medfødt lipodystrofi. J Clin Endocrinol Metab. 2008 April; 93 (4): 1129-34.
- Rajab a, Straub V, McCann LJ, et al. I en ny form for medfødt generaliseret lipodystrofi med muskelrippling (CGL4) på grund af ptrf-CAVIN mutationer. PLoS genetics. 2010 12. marts; 6 (3): e1000874.
- Payne F, Lim K, Girousse A, et al. Mutationer, der forstyrrer Kennedy phosphatidylcholinvejen hos mennesker med medfødt lipodystrofi og fedtleversygdom. Proc Natl Acad Sci U S A. 2014 Juni 17; 111 (24): 8901-6.
- Dyment DA, Gibson vægt, Huang L, et al. Bialleliske mutationer ved PPARG forårsager en medfødt, generaliseret lipodystrofi svarende til Berardinelli-Seip syndrom. Eur J Med Genet. 2014 Sep; 57 (9): 524-6.
- Garg A. klinisk gennemgang#: Lipodystrofier: genetiske og erhvervede kropsfedtforstyrrelser. Journal of clinical endocrinology and metabolism. 2011 November; 96 (11): 3313-25.
- Lima JG, Nobrega LH, de Lima NN, et al. Kliniske og laboratoriedata fra en stor række patienter med medfødt generaliseret lipodystrofi. Diabetol Metab Syndr. 2016; 8: 23.
- Lima GJ, Lima NN, Oliveira CF, et al. Umbilical brok hos patienter med Berardinelliseip syndrom: er det virkelig brok. J Clin Mol Endocrinol. 2015; 1(1): 3.
- brun RJ, Araujo-Vilar D, Cheung PT, et al. Diagnosen og håndteringen af Lipodystrofisyndromer: en retningslinje for praksis i flere samfund. J Clin Endocrinol Metab. 2016 Dec; 101 (12): 4500-11.
- Lima JG, Nobrega LH, Lima NN, et al. Knogletætheden hos patienter med Berardinelli-Seip medfødt lipodystrofi er højere på trabekulære steder og hos type 2-patienter. J Clin Densitom. 2016 November 25.
- Simha V, Garg A. fænotypisk heterogenitet i kropsfedtfordeling hos patienter med medfødt generaliseret lipodystrofi forårsaget af mutationer i AGPAT2-eller seipin-generne. J Clin Endocrinol Metab. 2003 Nov; 88 (11): 5433-7.
- Musso C, Cochran E, Javor E, et al. Den langsigtede virkning af rekombinant methionyl human leptinbehandling på hyperandrogenisme og menstruationsfunktion i kvindelig og hypofysefunktion hos mandlige og kvindelige hypoleptinemiske lipodystrofiske patienter. Stofskifte. 2005 februar; 54 (2): 255-63.
- Jiang M, Gao M, Vu C, et al. Mangel på testikel seipin forårsager teratosoospermi syndrom hos mænd. Proc Natl Acad Sci U S A. 2014 13. Maj; 111 (19): 7054-9.
- Mitchell O, Feldman DM, Diakov M, et al. Patofysiologien af trombocytopeni ved kronisk leversygdom. Hepat Med. 2016; 8: 39-50.
- Joubert M, Jagu B, Montaigne D, et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes. 2017 Apr; 66(4): 1030-40.
- Lima JG, Lima NN, Lima RLM, et al. Glargine U300 Insulin as a Better Option than Degludec U100 to Treat a Congenital Generalized Lipodystrophy Patient. Clin Diabetes Res. 2017; 1(1).