Autosomal recessiv medfødt Ichthyosis / Actas Dermo-Sifiliogrrificas
introduktion
den seneste konsensusklassificering af ichthyosis skelner mellem 2 hovedformer: de ikke-syndromiske former, der kun præsenterer med hud manifestationer, og de syndromiske former, der også præsenterer med manifestationer i andre organer (tabel 1).1 blandt de ikke-syndromiske former identificeres 4 grupper: almindelige ichthyoser, autosomale recessive medfødte ichthyoser (ARCIs), keratinopatiske ichthyoser og andre mindre almindelige ichthyoser.Traditionelt blev gruppen af ARCIs opdelt i 2 lidelser, lamellær ichthyosis (LI) og medfødt ichthyosiform erythroderma (CIE). I den nye klassifikation blev Harlekin ichthyosis (HI) føjet til denne gruppe1 fordi inaktiverende mutationer i ABCA12-genet er blevet identificeret som ansvarlige for denne lidelse,2,3 mens nonsensmutationer i det samme gen kan give anledning til LI4 eller CIE5,6 fænotype. Andre mindre almindelige varianter inkluderet i gruppen af ARCIs er selvhelbredende collodion baby (SHCB), acral SHCB og badedragt ichthyosis.7-9
Konsensusklassificering baseret på de kliniske træk ved Ichthyosis1.
| Nonddromiske former | syndromiske former |
| ichthyosesichthyosis vulgarisrecessiv ichthyosis (nonsyndromic )ajimajor formerharlekin ichthyosisLamellar ichthyosiform erythrodermaMinor formerselvhelende collodion babyAcral selvhelende collodion babybadedragt ichthyosiskeratinopatisk Ichthyosestore formerepidermolytisk Ichthyoseoverfladisk epidermolytisk ichthyosisminor formsannulær epidermolytisk ichthyosiskurth-Macklin ichthyosisautosomal recessiv epidermolytisk ichthyosisepidermolytisk nevusandre Formerloricrin keratodermaErythrokeratodermia vararabilispeeling hudsyndrommedfødt retikulært ichthyosiform erythrodermaKLICK syndrom | syndromisk røntgenbundet Ichthyosrecessiv ichthyosis (syndromisk)ichthyosis follicularis, alopecia og fotofobi (IFAP) syndromconradi-Scammann-happle syndrom (chondrodysplasia punctata type 2)syndromisk autosomal ichthyosishudforstyrrelsernetherton syndromichthyosis-Hypothrichosis syndromichthyosis-skleroserende cholangitis syndrometrichothystrophyneurological lidelsersj-Larsson syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, mental retardation, enteropati, døvhed, perifer neuropati, ichthyosis, keratoderma.
der foreligger kun begrænsede data om Arcis ‘ epidemiologi. I USA er en prævalens ved fødslen på 1 pr.100 000 indbyggere for LI og på 1 pr. 200 000 indbyggere for CIE blevet estimeret. Andre undersøgelser har rapporteret en kombineret prævalens for LI og CIE på 1 pr.200 000 til 300 000 befolkning.10,11 i nogle lande som Norge er den estimerede prævalens større (1 pr.91 000) på grund af grundlæggermutationer.12 fundet af 1 eller flere tilbagevendende mutationer i en population kan skyldes, at mutationen forekom på et givet tidspunkt i historien og derefter blev overført fra generation til generation (grundlæggermutation), eller fordi regionen af genomet, hvor mutationen findes, har en DNA-sekvens, der er modtagelig for mutation (mutation hotspot). I Spanien er den anslåede forekomst af ARCI 1 ud af 138 000 i den almindelige befolkning og 1 ud af 61 700 blandt børn under 10 år.13 i visse regioner i Spanien kan forekomsten være endnu højere. På den galiciske kyst blev der f.eks. rapporteret om en prævalens på 1 pr. 33 000, hvilket også skyldtes en grundlæggereffekt.14
lamellær Ichthyosis og medfødt ichthyosiform Erythrodermakliniske egenskaber
selvom det oprindeligt blev antaget, at LI og CIE var forskellige enheder, har der været rapporter om patienter med mellemliggende kliniske manifestationer, og begge tilstande kan være forårsaget af mutationer i det samme gen.15,16 derudover kan patienter med den samme mutation, selv inden for samme familie, udvikle forskellige fænotyper.12,15
de fleste patienter fødes indhyllet i en kollodimembran, der gradvist forsvinder i de første uger af livet og erstattes af den endelige fænotype (Fig. 1A). Hypohidrose, svær varmeintolerance og negledystrofi ses ofte i både LI og CIE.17-19 patienter med LI har normalt mere alvorlige kliniske manifestationer end dem med CIE. De har store platelignende skalaer, ofte af en mørk farve, der dækker hele kroppens overfladeareal. Erythroderma er enten fraværende eller minimal. Sådanne patienter har normalt ectropion og til tider eclabium, hypoplasi af led-og næsebrusk, ardannelse alopeci, især ved kanten af hovedbunden og palmoplantar keratoderma (Fig. 1B og C). CIE er kendetegnet ved tilstedeværelsen af erythroderma og fin hvidlig skalering (Fig. 2). Nogle patienter har markeret erytem og generaliseret skalering. Vægten kan være stor og mørk farvet, især på ekstensor overflader af benene. I mindre alvorlige tilfælde er erytem mild, og skaleringen er fin.

kliniske træk ved lamellær ichthyose. En, brunlig lamellær afskalning. B, markeret plantar hyperkeratose. C, ardannelse alopeci i hovedbunden.
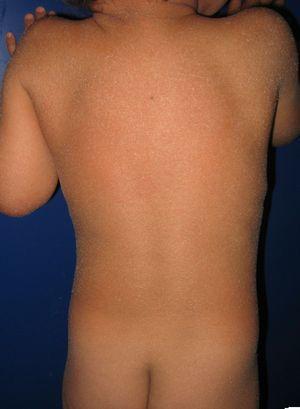
Patient med medfødt ichthyosiform erythroderma og mutationer i ALOKSE3-genet. Mild erytem og generaliseret hvidlig furfuraceous afskalning kan ses.
histopatologi
histopatologiske ændringer giver ikke en diagnose. I LI observeres massiv orthokeratotisk hyperkeratose, normalt med dobbelt forlængelse som i CIE. Epidermis er acanthotic og lejlighedsvis tager på en psoriasis-lignende udseende. Celleproliferationshastigheden er normal eller let forhøjet.17-19 patienter med CIE har mindre markant hyperkeratose med fokal eller omfattende parakeratose, et normalt eller fortykket granulært lag og mere udtalt acanthosis. Den epidermale omsætning øges.17-19
ultrastruktur
selvom der hidtil ikke er fundet en tæt sammenhæng mellem molekylære, kliniske og ultrastrukturelle fund, kan elektronmikroskopi ikke desto mindre være nyttigt til at udelukke andre former for ichthyose og til at styre genetiske analyser i nogle tilfælde. Fire typer medfødt ichthyose er blevet beskrevet (tabel 2).
Ultrastrukturel klassificering af medfødte Ichthyoser.
| Type | hovedfunktion | andre funktioner | mutationer | kliniske manifestationer |
| 1 | fravær af ultrastrukturelle markører af ichthyosis type 2, 3 og 4 | lipiddråber eller ringe i stratum corneum (hyppigst)små keratohyalin granulervesikulære eller lobulære membranbelægningsgranuler | TGM1 (33.3%)ALOKS12B (2 tilfælde) | CIE |
| 2 | kolesterol kløfter i stratum corneum | fravær eller udtynding af cornified envelopesmå keratohyalin granulesLipid dråber | TGM1 (89-100%) | LI |
| 3 | laminerede membranstrukturer i stratum granulosum og / eller stratum corneum. | unormal membranbelægning granulerlipid dråberfoci af fremtrædende sidestanukleære vakuoler i det granulære lag | NIPAL4 (93%) | CIE (hyppigst)LI |
| 4 | Trilamellære membranpakker, der fylder nogle celler i stratum granulosum og / eller stratum corneum | unormale membranbelægningsgranuler | FTAP4 | Ichthyosis prematuritet syndrom (100%) |
forkortelser: CIE, medfødt ichthyosiform erythroderma; LI, lamellær ichthyosis.
medfødt Ichthyosis Type 1
medfødt ichthyosis type 1 er kendetegnet ved fraværet af ultrastrukturelle markører for ichthyosis type 2, 3 og 4. Derfor stilles diagnosen normalt kun, når de andre typer er udelukket. Det hyppigste fund er tilstedeværelsen af lipiddråber eller ringe i stratum corneum (Fig. 3A).20 Disse lipiddråber er ikke et konstant træk eller specifikt for denne særlige type,da de ikke er til stede i alle tilfælde, 20 og de kan være til stede i andre typer ichthyosis.21,22 Klinisk er de fleste patienter til stede med manifestationer af CIE.12,20 en tredjedel af patienterne har mutationer i tgm1-genet.16 Denne ultrastrukturelle type er også blevet identificeret i forbindelse med mutationer i ALOKS12B-genet.23,24

Elektronmikroskopbilleder. A, medfødt ichthyosis type 1, der viser lipiddråber i stratum corneum og fravær af ultrastrukturelle markører af de andre typer ichthyosis. B, medfødt ichthyosis type 2, kendetegnet ved tilstedeværelsen af kolesterolklemmer (pil) i corneocytter.
medfødt Ichthyosis Type 2
medfødt ichthyosis type 2 er kendetegnet ved kolesterolklemmer i stratum corneum (Fig. 3B).21 sådanne kløfter er et konstant fund i denne type ichthyose og kan påvises i forskellige biopsier hos den samme patient; behandling med orale retinoider har ingen indflydelse på disse kløfter.12,25 Elektrontætte aggregater er også blevet observeret på corneocytter hos nogle patienter med mangelfuld TGase 1-aktivitet.26-28 klinisk har de fleste patienter alvorlige manifestationer af CIE.12 Denne ultrastrukturelle type er stærkt forbundet med mutationer i TGM1-genet.12,16
medfødt Ichthyosis Type 3
medfødt ichthyosis type 3 er kendetegnet ved lamellære membranøse strukturer i stratum granulosum og/eller stratum corneum. Disse strukturer er arrangeret i strimler omkring et tomt rum tæt på kernen.22,29-31 de kliniske manifestationer i denne type er forskellige fra de andre; begyndelsen af ichthyosis er variabel, afskalning og erytem kan være ujævn eller generaliseret, og især bøjningerne påvirkes. Mutationer i NIPAL4-genet er ansvarlige for 93% af ichthyoses type 3.32
medfødt Ichthyosis Type 4
karakteristisk er nogle celler i stratum granulosum og stratum corneum i medfødt ichthyosis type 4 fyldt med trilamellære membranpakker.33 disse fund er patognomiske for ichthyosis prematuritetssyndrom, en tilstand, der i øjeblikket betragtes som en syndromisk form for ichthyosis.34,35
molekylære undersøgelser
i genetiske termer er ARCIs meget heterogene. TGM1-genet er forbundet med de fleste tilfælde, men mutationer i 5 andre gener (ALOKS12B, ALOKS3, NIPAL4, CYP4F22 og ABCA12) er blevet rapporteret. Fischer et al.36 undersøgte 520 familier med ARCI og identificerede mutationer i mindst 1 af disse gener i 78% af tilfældene (TGM1 hos 32%, NIPAL4 hos 16%, ALOKS12B hos 12%, CYP4F22 hos 8%, ALOKS3 hos 5% og ABCA12 hos 5%). I en anden undersøgelse af 250 patienter med ARCI af forskellig oprindelse havde 38% tgm1-mutationer, 6,8% havde ALAKSE3-mutationer og 6,8% havde ALAKSE12B-mutationer.37 i Galicien identificerede vi mutationer i generne TGM1, ALOKS12B, ALOKSE3, NIPAL4 og CYP4F22 i 75% af de undersøgte familier, men fordelingen af mutationer var forskellig.14 TGM1-genet blev muteret i 68.7% af tilfældene, mens ALOKSE3-genet blev muteret hos kun 1 patient. Vi opdagede ikke mutationer i nogen af de andre 3 undersøgte gener.
TGM1
TGM1-genet er placeret på kromosom 14k11.2 og har 15 eksoner (GenBank NM-000359.2). Det koder for tgase 1, som er en af de 3 TGase, der findes i epidermis.38 dette deltager i dannelsen af den cornified kuvert ved at katalysere calciumafhængig tværbinding af flere proteiner, såsom involucrin, loricrin og prolinrige proteiner.39,40 det katalyserer også binding af ??- hydroksyceramider i det ydre lag af den cornified kuvert med proteiner i det indre lag.41,42 hos patienter med tgm1-mutationer mangler den cornified konvolut, og tgase 1-aktivitet er reduceret eller ikke-eksisterende.43-47
siden 1995, da dette gen blev identificeret som ansvarlig for nogle tilfælde af ARCI,er der rapporteret 48-50 mere end 110 mutationer hos patienter af forskellig oprindelse. Mutationer i TGM1 er den mest almindelige årsag til ARCI.36,37 denne mutation er fundet i 55% af tilfældene i USA og i 84% af tilfældene i Norge.12,51 den hyppigste mutation er c.877-2a>G, som er fundet i 34% af de muterede alleler, der er rapporteret til dato.52 den høje frekvens af denne mutation i lande som USA og Norge skyldes en grundlæggereffekt.12,53 den næsthyppigste mutation er S. Arg142His. Denne og lignende mutationer er blevet rapporteret i lande som Egypten, Tyskland, Finland og USA, 15,49-51,54-56,og det ser ud til, at disse er hotspot-mutationer.57 p. Arg307Trp-mutationen er hyppig i den japanske befolkning.5 i Galicien, p.Arg760, c.1223_1227delacaca og c.984 + 1g>a-mutationer i TGM1 blev identificeret i 81,82% af familierne med mutationer i dette gen, hvilket antyder en grundlæggereffekt.14 bekræftelse af denne hypotese blev opnået ved haplotype undersøgelse (arbejde endnu ikke offentliggjort).
TGM1 mutationer er ansvarlige for de fleste tilfælde af LI15,27,44,46,56,58-63 og for en lille procentdel af tilfælde af CIE.43,47,64,65 sådanne mutationer kan også give anledning til andre former for ARCI, såsom SHCB, acral SHCB og badedragt ichthyosis.
mange undersøgelser har forsøgt at demonstrere genotype-fænotypeforeninger mellem mutationer i TGM1 og ultrastrukturelle eller kliniske fund, men der er ikke observeret nogen signifikant korrelation til dato.15,16,53 generelt er patienter med mutationer i TGM1-genet mere alvorligt påvirket end dem uden sådanne mutationer. I en undersøgelse af 83 patienter med ARCI i Sverige og Estland var tilstedeværelsen af ectropion og collodion baby forbundet med tgm1-mutationer, mens en højere erytemhastighed blev observeret hos patienter uden mutationer i dette gen.66 en anden undersøgelse viste, at typen af skalering er den største forskel mellem bærere og ikke-bærere af TGM1-mutationer ved at finde ud af, at alle patienter med mutationer i dette gen havde lamellær skalering, mens 80% af dem uden TGM1-mutationer havde fin skalering.14 derudover er det blevet set, at trunkerende mutationer hyppigere er forbundet med hypohidrose og svedeforstyrrelser end missense-mutationer.51 i den nordamerikanske befolkning forudsiger en model baseret på tilstedeværelsen af visse kliniske egenskaber, at patienter, der er født som kollodionbørn og har okulære lidelser og/eller alopeci, er 4 gange mere tilbøjelige til at have TGM1-mutationer.51
ALOKSE3 og ALOKSE12B
alokse3-og ALOKSE12B-generne er placeret på kromosom 17p13.1.67 de har en lignende struktur med 15 eksoner, der koder for den epidermale Loks eloks-3 og 12R-loks.68,69 det faktum, at de overvejende udtrykkes i de suprabasale lag af epidermis, understøtter deres rolle i avancerede faser af epidermal differentiering med deltagelse i behandlingen af lamellære legemer.24,70 disse stoffer virker på tilstødende trin i hepoksilinvejen (Fig. 4). 12R-loks omdanner arachidonsyre til 12R-hydroksikosatetraensyre,mens eloks-3 omdanner dette produkt til en epoksialkoholisomer69, 71 af hepoksilin A3-familien.72 produktet er ustabilt og hydrolyseres i celler til et specifikt trihydroksid-derivat. Selvom den nøjagtige rolle af produkterne fra hepoksilinvejen ikke er kendt, er det blevet spekuleret i, at de kan deltage i dannelsen af intercellulære lipider i stratum corneum eller fungere som signaler til inducering af keratinocytdifferentiering.
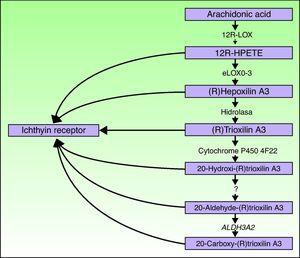
skematisk diagram over hepoksilinvejen, der viser deltagelsen af generene ALOKSE3, ALOKSE12B, NIPAL4 og CYP4F22. Mutationer i disse gener er ansvarlige for nogle typer ARCI. HPETE indikerer hydroperoksicosatetraensyre.
12B-og ALOKSE3-generne blev først identificeret i 2002,73, 74 siden da er der rapporteret mere end 30 mutationer i ALOKSE12B-genet23,24,37,75-77 og ca.10 i ALOKSE3-genet37,74,75. Disse mutationer er ansvarlige for 14% til 17% af ARCIs36,37 og 72.2% af SHCBs.23,78,79 det forårsagende forhold mellem disse mutationer og fænotype blev bekræftet ved at påvise,at den katalytiske aktivitet af epidermal LOKSEN blev fuldstændigt afskaffet hos patienter med disse mutationer75, 80 og ved anvendelse af dyremodeller, der reproducerede den ichthyosiforme fænotype set hos mennesker.81-83 begge gener er ansvarlige for en lignende procentdel af ARCI-tilfælde. Imidlertid er rækkevidden af forskellige mutationer i ALOKSE3-genet begrænset på grund af overvejelsen af 2 mutationer, s.Arg234h og S.Pro630Leu, som synes at svare til hotspots.37,74,75
patienter med mutationer i generne ALOKSE3 og ALOKSE12B viser normalt en Cie-fænotype.74,75,77 sværhedsgraden af skalering er mild eller moderat, og skalaerne har en hvidlig eller lysebrun farve. Erytem kan også være til stede. Så mange som 76% af patienterne er født som collodion babyer og 88% har svedforstyrrelser.37 patienter med mutationer i ALOKSE12B-genet viser mere begrænset, hvidlig afskalning sammenlignet med bærere af mutationer i ALOKSE3-genet. I disse tilfælde er vægten brunlig og klæbende. Tilstedeværelsen af erytem, palmoplantar hyperkeratose og accentuering af palmoplantarfoldene er også forbundet med aloks12b mutationer.37
Ichthyin/NIPAL4
NIPAL4-genet, også kendt som ichthyin-genet, er placeret på kromosom 5k33. Det har 6 eksoner, der koder for et protein med flere transmembrane domæner med ukendt funktion.84 det er blevet antaget, at proteinproduktet deltager i den samme metaboliske vej som loks og kan virke som en receptor for trioksiliner A3 og B3 eller for andre metabolitter af hepoksilins metaboliske vej.84 Det ville således være impliceret i dannelsen af lamellære legemer eller i deres transport mod det ekstracellulære rum.32 Til støtte for dette er 2 observationer. For det første er mutationer i dette gen i 93% af tilfældene forbundet med et ultrastrukturelt mønster af medfødt ichthyosis type 3, kendetegnet ved abnormiteter i de lamellære legemer og tilstedeværelsen af aflange perinukleære membraner i stratum granulosum.32 for det andet udtrykkes NIPAL4 i det væsentlige i epidermisens stratum granulosum, hvor de lamellære legemer er til stede.85
siden opdagelsen af NIPAL4-genet i 2004,84 er der kun rapporteret 9 mutationer hos patienter fra Middelhavslandene (Algeriet, Tyrkiet og Syrien), 84 skandinaviske lande,32 Pakistan,85 Færøerne,32 og Sydamerika.84
det kliniske spektrum af patienter med mutationer i dette gen er bredt, selv blandt medlemmer af samme familie. Mellem 3,7% 32 og 60% 84 er født som collodion babyer. Når kollodimembranen forsvinder, udvikler de fleste patienter manifestationerne af CIE med fine hvidlige skalaer på en erytematøs base i ansigtet og bagagerummet og større, brunlige skalaer på nakken, balderne og benene.84 markeret kserose, generaliseret brunlig retikulær hyperkeratotisk plak, der forekommer fremhævet i hudfoldene, og ansigtsdyschromi kan være til stede.32,85 derudover er palmoplantar keratoderma et hyppigt fund sammen med lejlighedsvise fingerkontrakturer og buede fingernegle. Nogle undersøgelser har rapporteret fund, der er mere typiske for LI.32,85 tilstedeværelsen af tegn og symptomer på atopisk dermatitis er rapporteret hos nogle patienter, skønt mutationer i FLG-genet ikke blev påvist i nogen af disse tilfælde.85
CYP4F22
flj39501 – eller CYP4F22-genet er lokaliseret på kromosom 19p13.12.86.Det har 12 eksoner87 og koder for et P450-cytokrom, familie 4, underfamilie F, polypeptid 2, homolog af leukotrien B4-hydro-hydroksylase (CYP4F2). Reaktionen katalyseret af produktet af FLJ39501 i huden og substraterne for denne reaktion kan udledes analogt med dets kendte homologer CYP4F2 og CYP4F3.88 det er blevet antaget, at CYP4F2 og CYP4F3 deltager i hepoksilinvejen ved at katalysere omdannelsen af trioksilin A3 til 20-hydroksi-(R)trioksilin A387, og at slutproduktet af denne vej, 20-carboksi-trioksilin A3, kan have en vigtig biologisk regulerende virkning i huden.89
til dato er kun 8 mutationer af dette gen blevet rapporteret i 12 sammenhængende familier fra Middelhavslande87 og i 1 familie af israelsk oprindelse.62
i de familier, der er rapporteret af Left Kursvre et al., 87 de fleste patienter havde en CIE-fænotype ved fødslen, og dette udviklede sig derefter til LI. patienter blev normalt født med markant erythroderma, dog uden nogen kollodimembran. Da de blev ældre, udviklede de generaliseret hvidgrå skalering, som var mere markant i det periumbiliske område, på balderne og på den nederste del af kroppen. Hyperlinearitet af palmer og såler og afskalning i hovedbunden, på tidspunkter af pityriasiform type, var hyppige.87 i en anden familie blev de 3 berørte medlemmer født som collidion babyer og udviklet intens erythroderma, generaliseret afskalning og palmoplantar keratoderma.62
ABCA12
i 2003 blev ABCA12-genet rapporteret at være ansvarlig for nogle tilfælde af LI og blev kortlagt til kromosom 2k34.4 Det blev efterfølgende bekræftet, at mutationer i dette gen også var ansvarlige for HI.2, 3abca12 koder for 53 eksoner og tilhører en familie af ABC-transportører, der binder adenosintrifosfat, samtidig med at det letter transporten af flere molekyler over cellemembranen.90 medlemmerne af ABCA-underfamilien er alle impliceret i lipidtransport.91 mangelfuld ABCA12-funktion forårsager lipidtransportforstyrrelser i lamellære legemer og fører således til et fald i intercellulære lipidniveauer i stratum corneum.3ultrastrukturelle undersøgelser har vist, at ABCA12 er placeret i lamellære legemer forbundet med glycosylceramider.91abca12-mutationer har været forbundet med forstyrrelser i distribution og transport af glycosylceramider og med nedsatte niveauer af hydroksyceramider, en af hovedkomponenterne i lipidbarrieren i de intercellulære rum.3,6,92,93 den massive hyperkeratose, der forekommer hos disse patienter, kan være et kompenserende respons på en mangelfuld lipidbarriere.94 det kan også skyldes manglen på afskalning af corneocytterne, 93 som kunne være forårsaget af defekter i transporten af visse proteaser, såsom callicrein 5 og cathepsin d, som følge af forstyrrelser i de lamellære legemer.95 Murine modeller og in vitro-undersøgelser antyder, at ABCA12-mutationer også har en effekt på epidermal differentiering.95-97
til dato er der rapporteret mere end 50 mutationer i ABCA12-genet hos patienter med ARCI fra Afrika, Europa, Pakistan og Japan. De hyppigste mutationer er p.Val244SerfsTer28,2,98,99 identificeret i pakistanske og indiske populationer og S.Asn1380Ser, 4 identificeret i afrikanske familier. I begge tilfælde kan disse være grundlæggende mutationer.
omfanget af abca12-mutationerne er relateret til fænotype, med mutationer forbundet med fuldstændigt tab af funktion, der fører til HI-fænotypen.2,3,98-102 derimod er de fleste mutationer i Li og CIE missense og har en mindre alvorlig effekt på proteinfunktionen.4-6, 103 mutationerne, der ligger til grund for LI-fænotypen, ser ud til at være koncentreret i det første adenosintrifosfatbindende kassetteområde.4 klinisk har patienter med CIE og mutationer i ABCA12-genet mellemstore skalaer, der er noget større end dem, der normalt observeres hos patienter med denne fænotype.
Harlekin Ichthyosis
hej eller harlekinfoster er en alvorlig og normalt dødelig form for ichthyosis. Børnene er normalt for tidlige med omfattende skinnende hyperkeratotiske plaketter, adskilt af dybe sprækker, der dækker hele integumentet og danner geometriske mønstre, der minder om tøj, der bæres af harlekiner, hvilket giver tilstanden sit navn. Hudtæthed fører til markant eversion af øjenlåg og læber, rudimentær udvikling af led-og næsebrusk og lejlighedsvis mikrocefali. Børnene har sjældent øjenvipper eller øjenbryn, selvom håret i hovedbunden kan bevares. Hænder og fødder er hævede og hævede og ofte dækket af et handskelignende lag. De kan have fingerkontrakturer.
for sådanne patienter er risikoen for at dø i nyfødtperioden meget høj.104 lungeventilation er kompromitteret; transepidermalt vandtab fører til dehydrering, vandkraft ubalance og termisk ustabilitet; og risikoen for infektioner øges. Tæthed i ansigtet og eclabium forhindrer sugning og derfor fodring med den tilsvarende forværring af dehydrering. Nyfødte med denne tilstand levede sjældent længere et par uger. I de senere år er chancerne for langvarig overlevelse imidlertid steget markant, hovedsageligt på grund af administration af systemiske retinoider og fremskridt inden for intensiv neonatal pleje.105 i en nylig undersøgelse overlevede 83% af patienterne behandlet med orale retinoider sammenlignet med 24% af de ubehandlede patienter. De fleste af dødsfaldene fandt sted i de første 3 dage af livet, men behandlingen blev først startet efter dette hos mange af de overlevende.104 dette antyder, at mange af disse tidlige dødsfald ville have fundet sted uanset retinoidbehandling.
de børn, der overlever den nyfødte periode, udvikler generelt alvorlig CIE.106 arten og placeringen af mutationer i ABCA12-genet og omfanget af transportørfunktionstab kan bestemme prognosen.3,92,107 patienter, der bevarer en vis grad af proteinaktivitet, omend minimal, kan have en bedre chance for at overleve. Bærere af homosygøse mutationer har en højere dødelighed.104
det vigtigste histologiske kendetegn ved HI er tilstedeværelsen af et ekstremt tykt og kompakt orthokeratotisk stratum corneum. Hårsækkene og svedkanalerne har fremtrædende hyperkeratotiske propper107,108 og har unormale eller fraværende lamellære kroppe, lipidindeslutninger eller rester af organeller eller kerner i corneocytterne og fravær af intercellulære lipider i den ultrastrukturelle undersøgelse.108.109 hårsækkene viser en markant koncentration af keratotisk materiale, hvilket er et diagnostisk træk ved HI, der anvendes til prænatal diagnose.
til dato er detektionshastigheden for mutationer i ABCA12-genet hos patienter med HI tæt på 100%, og det ser derfor ud til at være en genetisk homogen tilstand.
Collodion Baby og selvhelbredende Collodion Baby
Collodion babyer fødes normalt for tidligt, og perinatal sygelighed og dødelighed øges. Ved fødslen er nyfødte dækket af en skinnende undervist gennemsigtig membran, der minder om cellofanindpakning (Fig. 5). Babyer har ectropion, eclabiumog hypoplasi af nasal og ledbrusk. Sugning og lungeventilation kan hindres110 og transepidermalt tab af vand, og risikoen for infektioner øges.110,111

Collodion baby, der efterfølgende udviklede sig til en lamellær ichthyosis fænotype.
Collodion baby er den sædvanlige præsentation for HI og CIE. Autosomal dominant LI, 112,113 Sj Largren-Larsson syndrom,110 trichothyodystrophy,114 juvenil Gauchers sygdom, 110 neutral lipidlagringssygdom, Conradi-h Lutnermann-Happle syndrom, Hays-brønde syndrom og ektodermal dysplasi115 kan også lejlighedsvis præsentere som collodion baby. Membranen forsvinder spontant hos 10% til 24% af nyfødte for at give plads til helt normal hud.110.116 tidligere blev disse tilfælde beskrevet som li for den nyfødte, 117, men de kaldes ikke SHCB.118 nogle forfattere har foreslået udtrykket selvforbedrende collodion ichthyosis, fordi mange af disse patienter, når de undersøges senere i barndommen eller som voksne, har en variabel grad af anhidrose og varmeintolerance og milde tegn på ichthyosis, såsom kserose og fin afskalning, især i aksiller og nakke.78
hverken optisk mikroskopi eller ultrastrukturelle undersøgelser af collodion baby er specifikke. Det foretrækkes derfor at udsætte hudbiopsien, indtil den endelige fænotype er udviklet.
mutationer i tgm1,7,119alokse3,78 og ALOKS12B23,78,79 gener er blevet identificeret hos patienter med SHCB. ALOKS12B mutationer er de mest almindelige. I en serie på 15 skandinaviske patienter med SHCB havde 67% mutationer i ALOKS12B-genet, 25% i ALOKSE3-genet og 8,3% i TGM1-genet.78 mutationer blev ikke fundet hos nogle patienter, og derfor er andre gener sandsynligvis også impliceret. Der har været spekulationer om, at disse mutationer reducerer aktiviteten i livmoderen, men ikke efter fødslen.7 i livmoderen, hvor det hydrostatiske tryk er højt, omdanner chelation med vand det muterede ferment til en inaktiv konformation. Efter fødslen, når trykket falder, vender det tilbage til sin aktive form, og dets aktivitet øges tilstrækkeligt til at opretholde en normal eller minimalt påvirket fænotype.7
Acral selvhelbredende Collodion Baby
selvom collodion baby påvirker hele kroppen, er der rapporteret tilfælde begrænset til de akrale regioner. I 1952 Finlay et al.120 rapporterede et tilfælde af kollodimembran, der kun påvirkede hænder og fødder, og som fulgte et selvhelbredende kursus. For nylig er der rapporteret om et nyt tilfælde af acral SHCB i forbindelse med mutationer af TGM1-genet.8 Det vides ikke, hvorfor disse læsioner er begrænset til akrale regioner, selvom faktorer forbundet med stedafhængig regulering af aktivitet kan være i drift.8
badedragt Ichthyosis
badedragt ichthyosis blev først rapporteret som en uafhængig ARCI-variant i 2005, skønt der tidligere var rapporteret tilfælde af ichthyosis med en ejendommelig fordeling.121-123 det er hovedsageligt blevet påvist hos patienter med sydafrikansk oprindelse,9 skønt det også er rapporteret hos personer fra Europa og Middelhavslandene.124 ved fødslen har patienter en generaliseret kollodimembran, som derefter kaster for at forlade den karakteristiske fordeling af skalering. Stammen, den proksimale region af armene, inklusive aksillerne, nakken og hovedbunden påvirkes generelt, mens den centrale del af ansigtet, lemmerne og binyreområdet normalt Skånes.9 vægten er stor, lamellær og mørk i farven. Finere afskalning kan forekomme i popliteal og antecubital fossae.124.125 håndfladerne og fodsålerne har mild diffus hyperkeratose, mens ryggen på hænder og fødder ikke viser nogen involvering.
histopatologisk undersøgelse af påvirket hud viser markeret hyperkeratose uden parakeratose, normale granulære lag, mild eller moderat acanthosis og en mild lymfocytisk infiltration i den øvre dermis.9 elektronmikroskopi observationer er i overensstemmelse med medfødt ichthyosis type 2 i de fleste tilfælde. Ikke-involveret hud viser ingen unormale fund.124.125 i sund hud er tgase 1-aktiviteten lidt reduceret og sædvanligvis lokaliseret i pericellulære områder. I involveret hud er den resterende aktivitet og unormalt placeret i cytoplasmaet.124
mutationer er blevet påvist i TGM1-genet hos alle patienter med badedragt ichthyosis undersøgt til dato.119.124-126 den mest almindelige mutation er p.Arg315Leu, som er blevet identificeret hos de fleste Sydafrikanske patienter og kunne være en grundlæggende mutation. Oji et al.124 foreslog, at hudtemperaturen kunne spille en rolle i udviklingen af disse manifestationer. Ved hjælp af digital termografi viste forfatterne en stærk sammenhæng mellem kropstemperatur og afskalning, hvor de hotteste områder af kroppen var de mest berørte. Aufenvenne et al.127 viste et fald i optimal temperatur for tgase 1-aktivitet hos patienter med badedragt ichthyosis. Dette fald blev ikke observeret i sunde kontroller eller hos patienter med generaliseret LI. dette fald i temperatur ville forklare fænotypen af disse patienter. Den optimale temperatur er 37 liter C for det normale, men 31 liter C for det muterede.
behandling
det primære formål med behandling i ichthyosis er at eliminere skalering og reducere kserose uden at forårsage overdreven irritation (tabel 3). Inden der træffes beslutning om behandling, skal aspekter som patientens alder og køn, sygdommens type og sværhedsgrad samt omfanget og stedet for læsionerne tages i betragtning.128
terapeutisk strategi i autosomale Recessive medfødte Ichthyoser.
| terapeutisk strategi for autosomale recessive medfødte ichthyoses | |
| badning og mekanisk fjernelse af vægte | badning med natriumbicarbonat eller hvedestivelse, majsstivelse eller risstivelse; mekanisk fjernelse af skalaerne (1 eller 2 gange om dagen) |
| topisk behandling (sekventiel) | urinstofholdige fugtighedscremerkeratinolytika med propylenglycolkombineret keratinolytika (propylenglycol, urinstofsyrer eller urinstof)Keratinolytika kombineret med salicylsyretopiske retinoiderhos nyfødte og små børn, anvende et køretøj uden aktive ingredienser. Undgå urinstof, salicylsyre og mælkesyre på grund af risikoen for systemisk absorption |
| Oral behandling | orale retinoider (acitretin eller isotretinoin) |
| andre foranstaltninger | opfølgning af ectropion af oftalmologenregelmæssig rensning af det ydre øre ved øre-hals-næse specialistfysioterapi for at forhindre kontrakturer.Undgåelse af anstrengende aktiviteter i en høj omgivelsestemperaturhydroterapi |
badning og mekanisk eliminering af skalaer
daglig badning anbefales til patienter med ARCI for mekanisk at eliminere skalaer og spor af fugtighedscreme. Dette er lettere, hvis patienten er nedsænket i vand i 15 til 30minutter. Nogle forfattere anbefaler at tilsætte natriumbicarbonat til badet for at denaturere keratinerne og gøre vandet alkalisk og således lette eliminering af skalaerne.129 andre produkter, der kan tilsættes, inkluderer hvedestivelse, majsstivelse eller risstivelse. Badeolier er ikke egnede, da de kan føre til okklusion med efterfølgende risiko for bakterieproliferation og forværring af termoregulering.
topisk behandling
fugtighedscreme og topiske keratolytiske midler er normalt den første terapeutiske mulighed. De forbedrer hudens barrierefunktion og letter afskalning. Milde lokale bivirkninger, såsom forbigående kløe, irritation eller stikkende fornemmelse kan forekomme.
natriumchlorid, urinstof, vitamin E-acetat, glycerol og vaselin kan bruges som fugtighedscreme og smøremidler. Hos patienter med tyk skalering og markeret hyperkeratose kan der tilsættes 1 eller flere keratolytiske midler, såsom keratolytiske syrer (mælkesyre og glykolsyre),130 salicylsyre, N-acetylcystein,131-133 urinstof (>5%),134 og propylenglycol. Modulatorer af keratinocytdifferentiering anvendes også. Disse omfatter topiske retinoider (tretinoin, adapalen, tsaroten),135,136 calcipotriol,137 og dekspanthenol.Aktuelle retinoider forårsager ofte irritation og små, meget smertefulde sprækker.137 desuden er der risiko for absorption og teratogenicitet hos fertile kvinder, hvis de anvendes for meget.138 for at øge effektiviteten af keratolytika og fugtighedscreme kan okklusiv dressing påføres i specifikke områder, der er ildfaste til behandling.139 en additiv eller synergistisk virkning kan også opnås ved at kombinere 2 eller flere keratolytiske midler eller fugtighedscreme.140-142 behandling bør optimeres for hver enkelt person i betragtning af den meget variable karakter af tilstanden og hudfølsomhed og forskelle i respons på hver behandling. Optimeringsprocessen kan hjælpes ved at behandle den ene side af kroppen forskelligt fra den anden for at muliggøre sammenligninger. Nyfødte og små børn skal behandles med et køretøj uden aktive stoffer, da huden er meget fin og følsom, og de fleste keratolytika tolereres ikke. Derudover er risikoen for perkutan absorption af topiske produkter såsom urinstof, salicylsyre og mælkesyre større.143-145
systemisk behandling
orale retinoider har keratolytiske virkninger, der hjælper med at eliminere skalaer og forhindre overdreven hyperkeratose. Både isotretinoin og aromatiske retinoider (acitretin og etretinat) har vist sig effektive til behandling af ARCIs.128.146.147 Acitretin i en dosis på 0,5 til 1 mg/kg/D er det mest anvendte lægemiddel, især hos patienter med LI.148 patienter med CIE kan have et mere komplet respons og ved lavere doser.
de vigtigste bivirkninger er mukokutane lidelser, teratogenicitet, muskuloskeletale lidelser og unormal lipidprofil og forhøjelse af transaminase.149-152 med hensyn til teratogenicitet bør lægemidlerne i tilfælde af etretinat og acitretin undgås under graviditet, og patienter bør undgå at blive gravide i 3 år efter seponering af behandlingen.151 Isotretinoin har en kortere halveringstid og elimineres fuldstændigt fra organismen efter 1 måned og kan derfor være den foretrukne mulighed hos kvinder, der ønsker at blive gravid.128
behandlingsovervågning bør omfatte en laboratorieoparbejdning med en leverfunktionstest og lipidprofil, før behandlingen påbegyndes, derefter 1 måned og hver 3.måned efter behandlingsstart. Hos frugtbare kvinder skal der udføres en graviditetstest i de 2 uger, før behandlingen påbegyndes, og en effektiv antikonceptionsforanstaltning skal anvendes fra 4 uger før behandling til 3 år bagefter (i tilfælde af acitretin). Når langvarig behandling er påkrævet med retinoider, skal vækst og knogleudvikling overvåges. Nogle forfattere foreslår at udføre en knogleundersøgelse før behandling efterfulgt af en årlig undersøgelse.151 nylige retningslinjer anbefaler ikke at udføre rutinemæssig radiografi på grund af de mulige skadelige virkninger.152 i Stedet anbefales selektive radiografiske undersøgelser hos patienter, der har atypiske knoglesmerter.152
et alternativ til systemisk retinoidbehandling er brugen af lægemidler kendt som retinsyre metabolisme blokerende midler, som øger de endogene niveauer af retinsyre. Et sådant lægemiddel er liarosol, som har fået forældreløs status til behandling af LI, CIE og HI af Det Europæiske Lægemiddelagentur og US Food and Drug Administration.153-155 dette lægemiddel har vist sig at være mere effektivt end acitretin i kliniske forsøg, og det tolereres også bedre og har en bedre farmakokinetisk profil.154
anden medicinsk behandling
hos patienter med ectropion kan påføring af kunstige tårer og øjensmøremidler og især fugtgivende hud i ansigtet og kinderne reducere palpebral tilbagetrækning. Kirurgisk korrektion er en gyldig mulighed i alvorlige tilfælde, men dette skal normalt gentages et par år senere. Hydroterapi kan være gavnlig.156 patienter bør rådes til at undgå anstrengende fysisk aktivitet, når omgivelsestemperaturen er høj, da hypohidrose medfører risiko for heteslag og kramper. Orale retinoider kan forbedre termoreguleringen.157 fysioterapi er vigtig for at forhindre bøjningskontraktur, især i tilfælde af HI. Regelmæssig rensning af den eksterne auditive kanal af en øre-hals-næse specialist kan forhindre skalaer i at akkumulere og så forhindre høretab.
genetisk rådgivning og prænatal diagnose
når en patient diagnosticeres med ichthyosis, skal han eller hun tilbydes passende genetisk rådgivning, hvor arten af lidelsen, transmissionstilstanden og risikoen for fremtidige manifestationer i familien forklares. Prænatal diagnose kan indikere, om fosteret er påvirket, og hvis dette er tilfældet, kan psykologisk forberedelse af familien tilbydes og problemer forventes under graviditet og fødsel. Forældrene kan få mulighed for abort, hvis der ikke findes nogen behandling. Derudover skulle genterapi til disse tilstande blive tilgængelig i fremtiden, ville prænatal diagnose muliggøre anvendelse af denne terapi så tidligt som muligt.
i mere end 20 år blev prænatal diagnose udført ved at tage en biopsiprøve af føtalhud og studere den ved optisk mikroskopi, elektronmikroskopi eller immunhistokemi.158.159 denne invasive procedure kunne kun udføres i de sene faser af graviditeten mellem uge 15 og 23 af svangerskabet og var forbundet med en 1% til 3% risiko for at miste fosteret.160.161 identifikationen af de molekylære mekanismer for arvelige hudlidelser har muliggjort en meget tidligere diagnose baseret på genetiske teknikker.102.162-164 føtal DNA opnås ved fostervandsprøve udført mellem uge 15 og 20 eller ved chorionisk villusprøvetagning mellem uge 10 og 12. Risikoen for fostertab med disse teknikker er mindre end mellem 0,5% og 1%.165 andre ikke-invasive metoder under udvikling er analyse af føtal celle-DNA og frit føtal DNA i moderens cirkulation166 samt brugen af 3-dimensionel ultralyd.167.168
præimplantationsgenetisk diagnose kunne også være mulig i In vitro-befrugtningsteknikker, således at kun befrugtede æg, der er fri for mutationen, implanteres i livmoderen og derved undgår behovet for abort i de fleste tilfælde.169
fremtidige strategier for genetisk behandling af Ichthyosis
selvom der er gjort vigtige fremskridt med den genetiske diagnose af ichthyosis, forfølges der også nye strategier for disse sygdomme.170 huden er det mest tilgængelige organ til genoverførselsterapier, og sådanne teknikker er derfor minimalt invasive.171 imidlertid har huden også unikke immunologiske egenskaber, der ikke favoriserer langvarig ekspression af et transgent produkt.172 i LI lykkedes det en proces med tidligere vivo genoverførsel at genoprette normal tgm1-ekspression og korrigere fænotypen af hud transplanteret på bagsiden af immunsupprimerede mus.173.174 for nylig er fænotypen af dyrkede keratinocytter fra patienter med HI på grund af mutationer i ABCA12-genet også blevet genvundet.3
interessekonflikter
forfatterne erklærer, at de ikke har nogen interessekonflikter.