Frontiers in Neurology
introduktion
Neuroacanthocytosis er en overordnet betegnelse for en gruppe af sjældne syndromer, der er karakteriseret ved neurologiske symptomer i kombination med spiky deformerede røde blodlegemer (acanthocytter). Denne rapport fokuserer på Chorea-acanthocytose (ChAc) , som er en forældreløs sygdom med anslået 1.000 berørte individer over hele verden forårsaget af autosomal-recessive mutationer i vps13a (vacuolar protein sortering 13 homolog a) gen på kromosom 9K (1). Sygdommen kører et progressivt forløb, årsager til øget dødelighed kan være afvist motorfunktioner, der korrelerer med risikoforhold som dysfagi, men også pludselige uventede dødsfald er blevet beskrevet (2, 3). Indtil videre er der ingen årsagsbehandling.
den mistænkelige sameksistens af acanthocytose og bevægelsesforstyrrelser blev først rapporteret i 1970 ‘ erne af Levine og Critchley (4, 5) og er siden blevet accepteret som det fremtrædende træk ved sygdommen. Imidlertid beskriver vi med vores sagsrapport en sjælden klinisk fænotype af ChAc, der mangler åbenlyse bevægelsesforstyrrelser, hvilket antyder en bredere vifte af kliniske præsentationer. Diagnostiske værktøjer skal imødekomme disse udfordringer for at etablere den korrekte diagnose foreslår vi her molekylær neuroimaging som en nøglemetode.
sagsrapport
den mandlige indekspatient blev først præsenteret ved 25 år med to uprovokerede bilaterale toniske kloniske anfald. Der var ingen medicinsk historie. Hjernens MR-scanning og EEG i den alder var normale, og ingen behandling blev indledt på grund af patientens modvilje og sjældne hændelser. Imidlertid havde patienten igangværende anfald, der nu præsenterede sig som delvis epilepsi. Han beskrev gentagne følelser af pludselig svimmelhed, der blev diagnosticeret som en svimlende aura. Desuden havde han dyskognitive anfald, hvor han viste enten (a) manglende respons og recitering af tyrkiske bønner, (b) mundtlige automatismer eller (c) fikle med hænderne og udtale lyde—alt sammen delvist udviklet sig til toniske kloniske anfald. Video-EEG viste nu lokale spike-bølge komplekser både i højre og venstre temporal lap. I overensstemmelse med semiologien ved anfald blev diagnosen bilateral temporal lobepilepsi stillet, og medicinering med levetiracetam blev startet.
i en alder af 31 anfald blev ikke fuldstændigt undertrykt, som en del af yderligere diagnostik af temporal lobepilepsi gennemgik patienten en FDG-PET (Figur 1), som viste en bilateral mesiotemporal hypometabolisme i tråd med den mesiotemporale anfaldsoprindelse. Der var imidlertid også en markant, meget usædvanlig striatal hypometabolisme, der rejste mistanke om en neurodegenerativ bevægelsesforstyrrelse. Der blev derfor iværksat en omfattende opfølgningsevaluering (se figur 2). En FP-CIT-SPECT blev udført, hvilket gav et moderat bilateralt tab af striatal dopamintransportørtilgængelighed i tråd med et fald i nigrostriatal integritet (Figur 1). En MR med høj opløsning præsenterede nu bilateral atrofi af nucleus caudatus (figur 3). Neurologisk undersøgelse viste et diskret bundet gangmønster og hypomimi, men ingen anden kærlighed til det ekstrapyramidale motoriske system og ingen bulbar symptomer som fodring af dystoni. Bortset fra lav refleksstatus på underekstremiteterne, for hvilke nerveledningsundersøgelser bekræftede en sensorimotorisk aksonal polyneuropati, var den neurologiske undersøgelse normal. Neuropsykologiske symptomer omfattede aspekter af en angstlidelse, og patienten rapporterede vanskeligheder med kortvarigt hukommelsestab. Neuropsykologisk test bekræftede imidlertid ikke manifest mnestic-funktionsfejl, men snarere en uspecifik distraktion, som desuden kunne være blevet forbedret ved utilstrækkelig beslaglæggelsesundertrykkelse på det tidspunkt. Laboratorieundersøgelser var negative for enhver symptomatisk epilepsi (dvs., autoimmune-encephalitis antistoffer, antineuronale antistoffer). Cerebrospinalvæske viste ingen tegn på betændelse, men en moderat proteinforøgelse (765 mg/l). Analyse af biogene aminer i cerebrospinalvæsken afslørede en forhøjelse af glutamin, som vi forbandt med nylige anfald. I det perifere blod blev 0,4% acanthocytter fundet. Serologisk undersøgelse af sygdommen var negativ. Mærkbar var en vedvarende forhøjelse af kreatinkinase (interval: 1.000–3.000 U/l), hvilket førte til den mistænkte diagnose af en mitokondriel lidelse. For yderligere undersøgelser blev der udført en iskæmisk laktattest, som viste normale resultater. En muskelbiopsi af M. gastrocnemicus blev udført, men analyse af mitokondriefunktionen og luftvejskæden var normal.
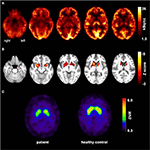
Figur 1. Resultater af molekylære billeddannelsesundersøgelser. Transaksiale FDG-PET-billeder (a) viste ikke kun en mild bilateral mesiotemporal hypometabolisme (i tråd med mesiotemporal anfaldsoprindelse), men også en markant striatal hypometabolisme, der viste sig at være meget signifikant sammenlignet med sunde kontroller . En yderligere FP-CIT-SPECT-undersøgelse (C) afslørede også et moderat tab af striatale dopamintransportører, der var vidne til et fald i nigrostriatal integritet (en aldersmatchet sund kontrol vises til sammenligning; DVR, fordelingsvolumenforhold).

figur 2. Klinisk og diagnostisk forløb for indekspatienten.
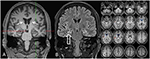
figur 3. MR viser diskret bilateral caudathovedatrofi og en mindre og hyperintense højre sidet hippocampus, der indikerer højre sidet hippocampal sklerose . Den venstre hippocampus er noget hyperintense, men ikke atrofisk. Bilateral caudathovedatrofi bekræftes med CVR-analyse, hvor blå farver viser nedsat grå og røde farver henholdsvis øget cerebrospinalvæskevolumen (C).
familiehistorie afslørede en sammenhæng mellem hans forældre (første fætre), som selv ikke havde nogen signifikant medicinsk historie. Af hans seks søskende havde to (i alderen 20-30 år) nu også lidt af generelle anfald (se figur 4). Lægejournaler over hans søskende var ikke tilgængelige for os.
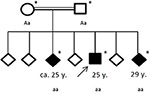
figur 4. Stamtavle af den berørte familie, med alder af første epileptiske anfald i år (y). Pil: indeks patent. Asterisk: genetisk test udført. Aa, heterosygøs; aa, homosygøs for c. 4326 T>A (S.Tyr1442*).
under hypotesen om en arvelig neurodegenerativ bevægelsesforstyrrelse blev patienten og hans familie henvist til genetisk testning. Eksomsekventering afslørede en homosygøs roman trunkerende mutation c.4326 t>A (S.Tyr1442*) i ChAc-genet VPS13A hos de tre berørte søskende. De sammenhængende forældre blev begge opdaget heterosygøse. Ingen andre varianter eller mutationer blev identificeret i eksomsekventeringen.
faktisk havde indekspatienten i en opfølgning i en alder af 33 år kun marginale ekstrapyramidale symptomer, og ingen var til stede i nogen af hans søstre. Han gav nu indtryk af let stivhed kun under priming på højre øvre lem og en mild bradykinesi i de øvre lemmer. Under behandling med levetiracetam og lacosamid har patienten været anfaldsfri.
Diskussion
med det foreliggende tilfælde demonstrerer vi, at ChAc klinisk kan manifestere sig med temporal lobepilepsi, der mangler nogen signifikante motoriske symptomer på grund af en ny tilsvarende mutation i VPS13A-genet.
sygdomsforløbet er typisk kendetegnet ved en progressiv bevægelsesforstyrrelse (inklusive chorea, dystoni, parkinsonisme), kognitive og psykiatriske ændringer og myopatiske symptomer med serologiske markører for acanthocytose og hyperckæmi. Krampeanfald er tidligere blevet bemærket som et symptom på ChAc (6), men bevægelsesforstyrrelser betragtes stadig som det vigtigste symptom. Der opstår bevis for, at epilepsi, og mere specifikt epilepsi med oprindelse i den temporale lap, kan være en undervurderet fænotype af ChAc. Peluso et al. rapporterede en patient svarende til vores, som selv i en alder af 46 år ikke viste bevægelsesforstyrrelser, mens de præsenterede neuroimaging-fund, der tyder på basal ganglier involvering (7). I overensstemmelse med det, Scheid et al. beskrevet tre patienter, der led af mesial temporal lobepilepsi og sklerose (baseret på MR-undersøgelser) som det dominerende symptom og blev diagnosticeret med ChAc (8). Desuden Mente et al. for nylig leverede obduktionsresultater af en ChAc-patient, der ikke kun viste basalganglieratrofi, men også hippocampal sklerose (9). I tråd med vores sag synes bilateral involvering af hippocampus at være et almindeligt fund (10, 11). Den nøjagtige sammenhæng mellem anfald og sklerose er dog stadig et spørgsmål om høne eller æg. Choreins korrelerende rolle i den strukturelle udvikling af hippocampus er endnu ikke fuldt ud forstået. Interessant nok blev der i en musemodel af ChAc set strukturelle ændringer, da hippocampal proteinekspression af GABA (A) receptor gamma 2 og dets forankringsprotein chorein blev forøget (12).
i klinisk rutine kan denne fænotype stadig være udfordrende for at finde den korrekte diagnose. Det første afgørende tip mod en neurodegenerativ sygdom hos vores patient var et fremtrædende fald i striatal glukosemetabolisme og et moderat fald i nigrostriatal integritet på FDG-PET og FP-CIT-SPECT, henholdsvis. Den lille samling af funktionelle billeddannelsesresultater, baseret på sagsseriebeskrivelse, viser, at ændringer i metabolisme og dopaminerg dysfunktion for det meste forekommer i caudatkernen og putamen (13). Hjerne MR afslørede atrofi af nucleus caudatus i løbet af sygdommen, som er blevet beskrevet som et typisk fund i ChAc (14). Følgelig inkluderer forskellige neurologiske træk normalt chorea, fodringsdystoni, orofaciolingual dyskinesier og tics sammen med andre symptomer på basalganglier, såsom parkinsonisme og dystoni (3). Det er fristende at spekulere i, at de observerede striatale ændringer i vores patient stadig var under tærsklen for at forårsage symptomer (dvs.prodromale billeddannelsesresultater), som i sidste ende kan forekomme, når sygdommen skrider frem. Vi opfordrer derfor til strukturel og molekylær billeddannelse som et følsomt diagnostisk værktøj i denne forældreløse sygdom.
i ChAc er der beskrivelser af acanthocytantal mellem 5 og 50% (3), men der er et par sagsrapporter, der viser, at acanthocytter kun kan forekomme sent i sygdomsforløbet (15), kan være meget lave eller endda være helt fraværende (16). Derfor bør acanthocytter ikke betragtes som obligatoriske for at stille diagnosen. Derudover kan epilepsi forsinke diagnosen, da den skjuler andre typiske symptomer på ChAc. Især kan psykiatriske symptomer såsom personlighedsændringer og kognitiv forringelse, som er meget almindelige, misforstås og tilskrives lægemiddelrelaterede bivirkninger (dvs.af levetiracetam) eller anfaldsrelaterede. Sekundær hyperckæmi ses efter generelle toniske kloniske anfald, og en vedvarende forhøjelse kan overses.
vores sag fremhæver også den afgørende fordel ved eksomsekventering for at etablere en korrekt diagnose, især i sjældne sygdomme, eller når symptomerne er vage. Familien af pattedyr VPS13 (A–D) gener er af stigende interesse, og mutationer er blevet identificeret i andre neurologiske sygdomme, såsom autosomal recessiv Parkinsons sygdom eller autosomal recessiv spinocerebellar ataksi (17). Recessive mutationer i VPS13D forårsager bevægelsesforstyrrelser i barndommen (18). Den underliggende patofysiologi af ChAc er endnu ikke fuldt dechiffreret, men det blev vist, at VPS13A koder for proteinet chorein som udtrykkes allestedsnærværende i hjernen og i et stort antal andre væv (19). Undersøgelser afslører, at det deltager i signalveje, der regulerer cytoskeletal arkitektur, eksocytose og celleoverlevelse (20). En anden mutation, der er beskrevet for at præsentere med en anfaldsdomineret fænotype hos 9 patienter med ChAc, er C.2343del-mutationen i VPS13A-genet (21). Det er rimeligt at antage, at specifikke mutationer i det samme gen resulterer i en vis aberration af chorein, der forårsager en unik fænotype, for eksempel ved at påvirke forskellige områder af hjernen. Den underliggende patofysiologi er dog stadig kun spekulativ, og yderligere dokumentation af de forårsagende mutationer vil være af interesse. Vi viser her, at den trunkerende mutation c.4326 T>A (S.Tyr1442*), som ikke er beskrevet før, resulterer i en lignende fænotype med overvejende epilepsi hos alle berørte familiemedlemmer.
afsluttende bemærkninger
epilepsi (især bilateral temporal lobepilepsi) er sandsynligvis en dominerende fænotype af ChAc. Derfor bør en søgning efter yderligere røde flag (såsom hyperckæmi, familiehistorie, polyneuropati) udføres omhyggeligt. I mistænkelige tilfælde skal søgning efter acanthocytter i blodet og hjernen MR tilføjes, da disse værktøjer er bredt tilgængelige. Hvis du er i tvivl, skal diagnostik suppleres med FDG-PET og/eller FP-CIT-SPECT, da de er følsomme til påvisning af striatal involvering. I autosomale recessive epilepsier med vejledende fund for en neurodegenerativ sygdom skal VPS13A enkelt gentest eller Chorein vestlig blot derefter bruges til at etablere den rigtige diagnose. Sidstnævnte bør også overvejes i andre neurodegenerative lidelser uden genetisk defineret etiologi.
etisk Erklæring
skriftligt informeret samtykke blev opnået fra patienten til deltagelse i undersøgelsen. Der blev indhentet skriftligt informeret samtykke fra patienten til offentliggørelse af denne sagsrapport.
Forfatterbidrag
JV, MR og SK deltog i patienthåndtering. JV skrev det første udkast til manuskriptet. LF, HU og PM udarbejdet tal. Alle forfattere bidrog til manuskriptrevision, læste og godkendte den indsendte version.
interessekonflikt Erklæring
HU er aktionær i Veobrain Gmbh, en spin-off af University Medical Center Freiburg.
de resterende forfattere erklærer, at forskningen blev udført i mangel af kommercielle eller økonomiske forhold, der kunne fortolkes som en potentiel interessekonflikt.
1. Det er en af de mest populære måder at gøre det på. Neuropsykiatri af neuroacanthocytose syndromer. Neuroci Biobehav Rev. (2011) 35:1275-83. doi: 10.1016 / j.neubiorev.2011.01.001
PubMed Abstrakt | CrossRef Fuld Tekst | Google Scholar
2. Rollator RH. Behandling af neuroacanthocytose syndromer. Tremor Anden Hyperkinet. Mov. (2015) 5:346. doi: 10.7916 / D8V66K48
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
3. A, Dobson-Stone C, Rampoldi L, Bader B, rollator RH, Danek A, et al. Chorea-Acanthocytose. Generevurderinger. Københavns Universitet (1993).
4. Critchley EM, Clark DB, A. Acanthocytose og neurologisk lidelse uden Betalipoproteinæmi. Arch Neurol. (1968) 18:134–40.
PubMed Abstrakt
5. Levine IM, Estes JV, Looney JM. Arvelig neurologisk sygdom med acanthocytose. Et nyt syndrom. Arch Neurol. (1968) 19:403–9.
PubMed Abstrakt | Google Scholar
6. Hardie RJ, Pullon HV, Harding AE, Oen JS, Pires M, Daniels GL, et al. Neuroacanthocytose. en klinisk, hæmatologisk og patologisk undersøgelse af 19 tilfælde. Hjerne (1991) 114 (Pt 1A): 13-49.
PubMed Abstrakt | Google Scholar
7. Peluso S, Bilo L, Esposito M, Antenora A, Rosa ADR, Pappata S, et al. Chorea-acanthocytose uden chorea: udvidelse af den kliniske fænotype. Parkinson Related Disord. (2017) 41:124–6. doi: 10.2016 / j. parkreldis.2017.05.013
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
8. Scheid R, Bader B, Ott DV, Merkenschlager a, Danek A. udvikling af mesial temporal lobepilepsi i chorea-acanthocytose. Neurologi (2009) 73:1419-22. doi: 10.1212 / VNL.0b013e3181bd80d4
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
9. Mente K, Kim SA, Grunseich C, Hefti MM, Crary JF, Danek A, et al. Hippocampal sklerose og mesial temporal lobepilepsi i chorea-acanthocytose: et tilfælde med klinisk, patologisk og genetisk evaluering. Neuropathol Appl Neurobiol. (2017) 43:542–6. doi: 10.1111 / nan.12403
PubMed Abstrakt | CrossRef Fuld Tekst | Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert a, la Foug Larre C, Noachtar S, et al. Bilateral temporal lobepilepsi bekræftet med intrakraniel EEG ved chorea-acanthocytose. Beslaglæggelse (2011) 20:340-2. doi: 10.1016 / j. beslaglæggelse.2010.12.007
PubMed Abstrakt | CrossRef Fuld Tekst | Google Scholar
11. Marson AM, Bucciantini E, Gentile E, Geda C. Neuroacanthocytosis: kliniske, radiologiske og neurofysiologiske fund i en italiensk familie. Neurol Sci. (2003) 24:188–9. doi: 10.1007 / s10072-003-0123-1
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
12. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Miesuno E, Kato M, et al. Choreinmangel fører til opregulering af gephyrin og GABA(A) receptor. Biochem Biophys Res Commun. (2006) 351:438–42. doi: 10.1016 / j. bbrc.2006.10.070
PubMed Abstrakt | CrossRef Fuld Tekst | Google Scholar
13. Ehrlich DJ, rollator RH. Funktionel neuroimaging og chorea: en systematisk gennemgang. J Clin Mov Disord. (2017) 4:8. doi: 10.1186 / s40734-017-0056-0
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
14. L-N, Lang AE. Neuropatologiske Fund i chorea-acanthocytose: ny indsigt i mekanismer, der ligger til grund for Parkinsonisme og anfald. Acta Neuropathol. (2014) 127:613–5. doi: 10.1007 / s00401-013-1241-3
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
15. Sorrentino G, de Renso A, Miniello S, Nori O, Bonavita V. sen udseende af acanthocytter i løbet af chorea-acanthocytose. J Neurol Sci. (1999) 163:175–8.
PubMed Abstrakt | Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Chorriso-acanthocytose uden acanthocytter. Revue Neurol. (2010) 166:100–3. doi: 10.1016 / j.neurol.2009.03.005
CrossRef Fuld Tekst | Google Scholar
17. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas a, et al. Tab af vps13c-funktion i autosomal-recessiv Parkinsonisme forårsager mitokondriel dysfunktion og øger PINK1/Parkin-afhængig mitophagy. Am J Hum Genet. (2016) 98:500–13. doi: 10.1016 / j.ajhg.2016.01.014
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
18. Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M, et al. Recessive mutationer i >VPS13D forårsager bevægelsesforstyrrelser i barndommen. Ann Neurol. (2018) 83:I6. doi: 10.1002 / ana.25204
PubMed Abstrakt | CrossRef Fuld Tekst | Google Scholar
19. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Miesuno E, Kato M, et al. In vivo distribution og lokalisering af chorein. Biochem Biophys Res Commun. (2007) 353:431–5. doi: 10.1016 / j. bbrc.2006.12.059
PubMed Abstrakt | CrossRef Fuld Tekst | Google Scholar
20. Lang F, Pelsl L, Sch Kursls L, Hermann A, F Kurllers M, Sch Kurtffer TE, et al. Neuroner, erythrocytter og videre -choreins forskellige funktioner. Neurosignaler (2017) 25:117-26. doi: 10.1159/000485457
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
21. Benninger F, Afa, kortsyn AD, Oliver KL, pendler M, Nakamura M, Et Al. Krampeanfald som præsentere og fremtrædende symptom i chorea-acanthocytose med C.2343del VPS13A genmutation. Epilepsi (2016) 57:549-56. doi: 10.1111 / epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar