Frontiers in Pediatrics
- introduktion
- langt KVT-syndrom
- 1
- LKTS2
- 3
- LKTS4
- LKTS5
- LKTS6
- LKTS7
- LKTS8
- LKTS9 og LKTS10
- LKTS11
- LKTS12
- LKTS13
- erhvervede LKT
- elektrokardiografiske træk i de tre almindelige former for langt KVT-syndrom
- genotype-fænotype
- klinisk behandling af hjertelidelser
- : Saudi Perspective
- interessekonflikt Erklæring
- anerkendelser
introduktion
familiær eller arvelig hjertearytmi omfatter betydelige procentdele af arytmier og også årsag til pludselig hjertedød (SCD) (1, 2). I løbet af de sidste to årtier har forskere og klinikere ydet en enorm indsats for at afsløre de indviklede og komplekse mekanismer ved medfødte familiære arytmier (1-8). For at forstå mekanismen for arytmogenese skal vi kende det grundlæggende i hjertecellulær struktur og deres elektrofysiologiske egenskaber. Hjertemyocytter er de vigtigste fungerende celler i hjertet, og de er i vid udstrækning koblet, så impulser formeres hurtigt og ensartet. Kardiomyocytter adskilles fra hinanden ved en specialiseret grænse kaldet den interkalerede disk; gap junction proteiner, hjerte desmosomer og ionkanaler er placeret i den interkalerede disk (9). Gap-kryds består af tætpakkede forbindelser, der tillader intercellulær udveksling af små molekyler og også tillader at strømme de spændende strømme fra en celle til dens nabocelle. Desmosomer sammen med adherens-krydsene er ansvarlige for de mekaniske vedhæftninger af individuelle kardiomyocytter. Alle disse komponenter i de interkalerede diske er sekventielt adskilt, og hver komponent udøver sin unikke funktion; forstyrrelse af den ene komponent påvirker funktionen af andre komponenter, som prædisponerer hjertet for at udvikle arytmier (9-11). Hjerteionkanaler er poreformende proteinkomplekser, der giver spænding gated og indviklet koordineret ind-og udadgående bevægelse af Ioniske strømme over cellemembranerne, afgørende for hjerterytmegenerering og udbredelse. Hjerteledningsdefekt (CCD), BrS, catecholaminerg polymorf ventrikulær takykardi (CPVT), tidligt repolariseringssyndrom (ERS) og familiær atrieflimren (af) er de i øjeblikket kendte hjertekanalopatier, som kan forekomme på grund af en enkelt eller multiple defekt i generne forbundet med hjerterytmegenerering og formering.
i denne gennemgang beskriver vi udelukkende på LKTS-forbundne arytmier, dens patofysiologi og i øjeblikket tilgængelig klinisk ledelse. Vi har været banebrydende for at belyse genetisk patologi i familiær hjertearytmi i Saudi-Arabien (1, 12-14), Vi gennemfører stadig kardiogenetiske undersøgelser i Saudi-Arabien, i slutningen af denne gennemgang vil vi diskutere den aktuelt tilgængelige viden om de genetiske og kliniske fund opnået fra flere Saudiarabiske familier med historie med synkope og SCD. Vi vil også tilføje de nyligt offentliggjorte data fra et team på King Faisal Specialist Hospital og Research Center, Riyadh (15).
langt KVT-syndrom
medfødt KVT er en arvelig lidelse defineret ved forlængelse af KVT-intervallet på elektrokardiogram (EKG). Patienter med alle former for hjertelidelser er disponeret for ventrikulær takyarytmi, torsades de pointes (TdP), der fører til tilbagevendende synkope eller SCD. I mange tilfælde kan synkope eller pludselig død være den første og den eneste manifestation. 1 ud af 2.000 mennesker verden over (16). Dette er en af de mest almindelige årsager til hjertelidelser i kroppen. Normale værdier for HTC er 440 ms hos mænd og 450 ms hos kvinder. Hos børn er alder og kønsafhængige værdier relevante. Den seneste konsensusanbefaling til diagnose er følgende (17, 18):
1.
a. i nærvær af en risiko score på 3,5 i fravær af en sekundær årsag til forlængelse af KT og/eller
b. i nærvær af en utvetydigt patogen mutation i et af KTS-generne eller
c. 500 ms i gentaget 12-bly elektrokardiogram og i fravær af en sekundær årsag til HT forlængelse.
2. En patient med uforklarlig synkope kan diagnosticeres i nærvær af en KTC mellem 480 og 499 ms i gentagne 12-bly EKG ‘ er i en patient med uforklarlig synkope i fravær af en sekundær årsag til KT forlængelse og i fravær af en patogen mutation.
patienter med KTC-varighed 500 ms havde en signifikant højere kumulativ Sandsynlighed for at opleve deres første synkope sammenlignet med patienter med KTC-varighed < 500 ms fra fødsel til alder 20 år (19). Men patienter, der har oplevet 2, 3 og 4 synkope episoder, var risikoen for en efterfølgende synkope episode næsten identisk mellem patienter, der havde smal eller langvarig KTC Varighed (19). Det molekylære grundlag er heterogent, og til dato er mutationer i 13 forskellige gener blevet beskrevet kausal til LKT ‘ er (1-8, 19, 20). En genetisk defekt findes normalt hos 70% af patienterne i et af disse 13 gener (1-8, 19, 20). Blandt de i øjeblikket kendte 13 forskellige typer LKT ‘er er de mest almindelige LKT’ ER1, LKT ‘ER2 og LKT’ er3 på grund af defekter i hjerteionkanalgener, KCNK1, KCNH2 og SCN5A, henholdsvis. Halvfems procent af alle kausale mutationer findes i disse tre gener (1-8, 19, 20). Mutationer i de resterende 10 gener er sjældne og udgør kun 10% af alle de nuværende kendte mutationer.
langt KT-syndrom er normalt en autosomal dominerende sygdom, men lejlighedsvis kunne flere mutationer i et enkelt gen eller i forskellige gener findes hos 5-10% af patienterne med KT ‘ er (21, 22). Patienter med flere mutationer kunne udvise en længere Kvtc sammenlignet med dem med en enkelt mutation, og sådanne patienter har også en 3,5 gange øget risiko for livstruende hjertehændelser (21, 22)
1
mutationer i KCNK1-genet er den mest almindelige form af alle LKT ‘er og kaldes type 1 (LKT’ er1). Halvtreds procent af alle kausale mutationer findes i kcnk1-genet (20). Kvlkv1 (også kaldet Kv7.1) er et protein fremstillet af kcnk1-genet, og proteinet danner tetramere i det endoplasmatiske retikulum inde i cellerne, som formodentlig samles sammen med proteinminken (kodet af KCNE1) og transporteres derefter til plasmamembranen i hjertemyocytterne, hvor de medierer en langsomt aktiverende strøm, der fremskynder repolariseringen af handlingspotentiale i hjertevæv, denne strøm er kendt som IKs (Figur 1). 1 kausale kcnk1-mutationer er for det meste missense-mutationer, og i sjældne tilfælde kan de være frameshift-mutationer i den C-terminale region (23). Hjertearytmi hos kcnk1-mutationsbærere udløses af adrenerge drev, f.eks. følelsesmæssig stress, fysisk anstrengelse, dykning, svømning (24-26). Patienter med mutationer ved transmembrane domæner af Kvlkv1 har højere risiko for hjertehændelser og har større følsomhed over for sympatisk stimulering (26, 27). Patienter med missense-mutationer har øget risiko sammenlignet med patienter med ikke-sense eller trunkerende mutationer (27, 28). Variation i 3 ‘ – UTR i kcnk1-genet påvirker også arytmifølsomheden signifikant, formodentlig ved at påvirke ekspressionen af genet (29). Patienter med arytmier på grund af kcnk1-mutationer reagerer ganske godt på kur-blokkere, men nogle patienter kan stadig være mindre lydhøre eller endda resistente over for denne medicin. I en nylig artikel, Barsheshet et al. (30) hævdede, at patienter med mutationer uden for det cytoplasmatiske loop (c-loop)-område i Kvlkv1 er mindre lydhøre over for LR-blokkere.
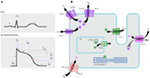
Figur 1. Ioniske strømme, der bidrager til det ventrikulære handlingspotentiale (A) og skematisk repræsentation af en kardiomyocyt, der (kun) viser de proteiner, der er involveret i patogenesen af arvelige arytmisyndromer (B). I (A) er handlingspotentialet justeret med dets omtrentlige virkningstid under EKG. I (B) er ankyrin-B, et adapterprotein involveret i det lange KT-syndrom type 4, ikke afbildet.
homosygøse eller sammensatte heterosygøse mutationer i kcnk1-genet, som kan forårsage den recessive form af sygdommen, jervell og Lange-Nielsen syndrom (JLNS), type 1 (31), er ret sjældne. Jlns-patienter lider også af alvorlige hjertearytmier og døvhed (31, 32). Patienter med kcnk1-mutationer, der er årsag til JLNS, har normalt ingen funktionelle IKs (28, 31-33). Det er muligt, at nogle patienter ikke kunne have døvhed på trods af at de havde homosygøse eller sammensatte heterosygøse mutationer i KCNK1, sådanne tilfælde kaldes autosomal recessiv LKTS1 (13, 32). Hos disse patienter kunne en lille mængde funktionel IKs-strøm (<10% af den samlede IKs) stadig være til stede, hvilket opretholder hørefunktionen, men deres hjerterytmefejl er lige så alvorlige som hos jlns-patienter (13, 32).
LKTS2
denne type LKTS er lige så udbredt som LKTS1, der tegner sig for 35-40% af LKTS-patienter med en påviselig mutation (1, 20, 24, 25). KCNH2 koder for HERG-proteinet (Kv11.1), som er den hurtigt aktiverende forsinkede ensretter k+ current (IKr). Patogene mutationer i dette gen, der reducerer Kv11.1-kanalfunktionen, forlænger varigheden af KVT-intervallet (Figur 1) og er årsag til KVKTS2. 2 forekommer under hvile / søvn, og kun 13% af de synkopale angreb blev rapporteret at forekomme under træning (25, 34). Pludselige overraskende lyde, f.eks. vækkeurringe, dørklokker, telefonringe, udløser typisk synkopale angreb hos disse patienter (24, 34). Patienter med mutationer i det poredannende område af Kv11.1 (kodet af kcnh2-genet) er modtagelige for høj risiko for arytmirelaterede hjertehændelser sammenlignet med patienter med ikke-poreregionsmutationer (35). Hos nyfødte er 2: 1 atrioventrikulær (AV) blok fortrinsvis forbundet med KCNH2 mutationer (36). Komplet AV-blok kompliceret af LKT ‘ er blev også fundet hos 17% af voksne patienter med en mutation i KCNH2-genet (37). Homosygøse mutationer i KCNH2 er sjældne, og når de er til stede, lider patienter af en alvorlig form for LKT ‘ er, med 2:1 AV-blok og alvorlige ventrikulære arytmier under intrauterine stadier såvel som efter fødslen (11, 38-40). Ud over de mange mutationer, der er beskrevet i LKTS2-patologi, kunne almindelige polymorfe varianter i KCNH2-genet også modulere sygdommens sværhedsgrad. Et spændende eksempel er k897t polymorfisme (SNP) i KCNH2, som er til stede i 33% af den generelle befolkning (41). K897T blev rapporteret at forværre patogeniciteten af kcnh2-mutationerne (42, 43).
3
Nav1.5 er den poreformende kur-underenhed af den spændingsafhængige hjerte-na+ – kanal, det er et integreret membranprotein kodet af SCN5A-genet og er involveret i initiering og ledning af hjerteaktionspotentialer. Hjerte-Na + – kanaler er sammensat af en poreformende karrusel-underenhed (kodet af SCN5A) og en eller flere ekstra karrusel-underenheder.
LKTS3 er forårsaget af forstærkning af funktionsmutationer, der forstyrrer hurtig inaktivering af larr-underenheden (Figur 1), og en mutation i SCN5A-genet er blevet beskrevet hos < 10% af alle LKT-patienter med en mutation (1, 20, 24). 3 patienter oplever størstedelen (39%) af deres hjertehændelser under søvn/hvile (25), og ca.13% af hændelserne blev rapporteret at forekomme under træning (25). I flere tilfælde blev en enkelt SCN5A-mutation vist at udøve to eller endda tre forskellige fænotyper af arytmier i samme familie, f.eks. Mandlige patienter med en lkts3-mutation kunne udvikle symptomer meget tidligere end de kvindelige patienter (48). Både heterosygøse og homosygøse SCN5A-mutationer er beskrevet i LKTS3 med funktionel 2: 1 AV-blok (49). Vi har for nylig fundet en iransk familie med mutationen 1507_1509 KKP hos flere familiemedlemmer, hvor patienter havde kombineret CCD og CCD (data ikke vist). Denne mutation er også rapporteret hos patienter fra andre lande, hvilket antyder, at dette er en tilbagevendende og hot spot-mutation . Mutationer med sådanne Tabs – og forstærkningsfunktionskarakteristika i forskellige faser af handlingspotentialet blev også rapporteret i 1493delk-og 1795insd-mutationer (44, 51).
lejlighedsvis kunne en SNP også udøve patogen virkning på dens bærere. S1103Y er en almindelig variant i SCN5A-genet, der findes i 13% af afroamerikanerne (52). Bærere med denne variant har øget risiko for arytmier og pludselig spædbarnsdødssyndrom (SIDS) (52).
LKTS4
LKTS4 repræsenterer den første ikke-kanalform af LKTS. En mutation i ANK2, et adapterprotein, fører til intracellulær calciumoverbelastning, som bidrager til LKTS4 (53, 54). Patienter med dette syndrom kan have sinusbradykardi, paroksysmal af og CPVT (54). Patogen effekt af ANK2-mutationerne kan være moderat til svær, og de kliniske udtryk afhænger af sværhedsgraden af mutationen.
LKTS5
mutationer i KCNE1 er forbundet med LKTS5 (Figur 1) (55, 56). Heterosygøse kcne1-mutationer reducerer IKs ved at udøve en dominerende negativ effekt på dens ledsagende normale allel og føre til forsinket hjerte-repolarisering (Figur 1), der er ansvarlig for øget risiko for arytmier (6). Patienter med homosygøse kcne1-mutationer lider af JLNS (type 2) (57, 58).
D85N er en polymorfisme i KCNE1-genet, der er til stede i 0.7-1% af den generelle befolkning (41). I en undersøgelse foretaget af Nishio et al. (59), blev d85n-polymorfisme hyppigere fundet hos patienter med LKT, hvilket gjorde det til en risikogenotype i LKT-patologi (potentielt kun i den asiatiske befolkning). I Europa blev D85N rapporteret hos 5% af de erhvervede patienter (i en kohorte på 32 patienter), som havde oplevet TdP (60).
LKTS6
KCNE2-gen koder for MinK-relateret peptid 1 (MiRP1), en formodet kar-underenhed af den kardiale Kaliumkanal IKr (Figur 1). Mutationer i kcne2-genet kan også føre til defekter i den hurtigt aktiverende komponent i den forsinkede ensretterkaliumstrøm (IKr), patologisk grundlag for KKTS6 (61). Auditiv / akustisk stimulus som vækkeur støj, dørklokke osv. kunne fremkalde synkopale angreb i kcne2-mutationsbærere, svarende til KCNH2-mutation (62).
LKTS7
dette syndrom er også kendt som Andersen–Tavil syndrom (ATS). ATS er en sjælden lidelse, manifesteret ved lejlighedsvis synkope og hjertestop. EKG-funktioner inkluderer forlængelse af mildt KVT-interval, unormale u-bølger, hyppig ventrikulær ektopi, tovejs ventrikulær takykardi (VT) og polymorf VT. Dette syndrom udviser også ekstrakardiale træk, f.eks. periodisk lammelse af skeletmuskulatur og udviklingsproblemer, såsom ganespalte, lavt indstillede ører, kort statur og udviklingsfejl i lemmerne (63). Størstedelen af klinisk diagnosticerede ATS-patienter blev rapporteret at have en mutation i KCNJ2 (63). KCNJ2 koder for en poreformende underenhed af indadrettet kaliumkanaler (IK1) (Figur 1) (64, 65).
LKTS8
også kendt som Timothy syndrom (TS), patienter viser alvorlig KVT-forlængelse på deres EKG ‘ er, hvilket kombineres med syndaktisk, skaldethed ved fødslen og små tænder i 100% af tilfældene og mindre penetrerende hjerte strukturelle misdannelser, mental retardering, autisme og ansigtsdysmorfe træk (66). Der er to undertyper: TS1 (klassisk) og TS2 (sjælden form).
TS2 er kardiologisk severere end TS1 (66, 67). TS2-patienter mangler også syndaktisk (67). Mutationer i underenheden af den L-type calciumstrøm (ICA-L), der koder for genet CACNA1C, fører til begge former for TS (LKTS8). Der er alternativt splejset, gensidigt udelukkende ekson – 8 i CACNA1C-genet, for klarhedens skyld er de navngivet som ekson-8 og ekson-8a. i hjerte og hjerne, hvor CACNA1C overvejende udtrykkes, findes ekson-8 i Karr 80% af mRNA-transkripterne, og ekson-8a er til stede Karr 20% transkripter (66). G406r mutation i ekson – 8a er kausal til klassisk form af TS (TS1) og G402S i ekson-8 blev rapporteret i severer i TS2. En ny tilføjelse til denne liste er Ala1473Gly mutation, som er blevet beskrevet i et TS-spædbarn med yderligere udvidet fænotype (68). Alle mutationer var enten de novo eller mosaik i moderselskabet, og alle resultatgevinster af funktion af ICA-l-kanalen (66-69).
LKTS9 og LKTS10
mutationer i CAV3 eller SCN4B giver forstærkning af funktion i sen INa, hvilket forårsager en LKTS3-lignende fænotype (70-74). De er kendt som LKTS9 (associeret med CAV3-mutation) og LKTS10 (associeret med SCN4B-mutation).
Caveolae er pænt beskrevet af Engelman et al. (72) som “små huler” i plasmamembranen. De er små ubelagte gruber og betragtes som stedet for vigtige dynamiske og regulatoriske begivenheder ved plasmamembranen (72, 73). Caveoliner er de vigtigste proteiner i caveolae, og caveolin-3 (kodet af Gen CAV3) findes specifikt i kardiomyocytter og skeletmuskelceller. Flere hjerteionkanaler er specifikt rapporteret at være lokaliseret i caveolae ekstraheret fra hjertemyocytter, der er beriget med caveolin-3 (72, 73). Derudover er komponenter i den karrus-adrenerge receptorsignalkaskade også til stede i caveolae-berigede membraner (72, 73).
SCN4B koder for nav-Lut4, som er en hjælpelit-underenhed af hjertenatriumkanalen. Indtil videre er der kun rapporteret om en mutation (L179F) i dette gen i en Meksikansk familie med flere berørte familiemedlemmer (74). Mutation i dette gen viste sig at resultere i forstærkning af funktionen af Nav1.5-strømmen (74).
LKTS11
i hjertet medieres sympatisk regulering af hjerteaktionspotentialvarighed (APD) ved aktivering af en purpur-adrenerg receptor (Purpur-AR), som kræver samling af AKAP9 (Yotiao) med iks-underenheden (Kvlkt1). Mutation i AKAP9 forårsager 11 (75). Til dato er der kun rapporteret om en mutation, S1570L, i AKAP9 (75).
LKTS12
Mutation i genet for LKT-1-Syntrofin (SNTN1) er årsag til lkts12. Mutation i dette gen fører til forstærkning af funktionen af hjertenatriumkanalen (Nav1.5), som er det patologiske grundlag for LKTS12 (76).
LKTS13
g-proteinkoblet, indadrettet kaliumkanalunderenhed (Kir3.4) er kodet af KCNJ5-genet. En tab af funktionsmutation i dette gen kan forårsage LKTS13 (77). Indtil videre er kun en mutation, G387R, blevet beskrevet i en kinesisk familie med ni patienter, der har denne mutation. Reduceret plasmamembranekspression af Kir3.4 blev foreslået som patologi af LKT ‘ er hos patienterne.
erhvervede LKT
ud over de medfødte LKT ‘er findes der også en anden variant af LKT’ er kendt som alkt ‘ er, som er forårsaget af faktorer og stoffer, der nedsætter kaliumstrømmen og forringer myokardiumets evne til at repolarisere. Velkendte tilstande er kvindelig køn, hypokalæmi og lægemidler, der hæmmer hjertekaliumkanaler (78-80). Et antal almindeligt ordinerede lægemidler kunne også fortrinsvis binde og blokere HERG-kanalen (Kv11.1, et protein kodet af KCNH2-genet) og disponerer for alkt ‘ er (79, 80). For nylig blev det vist, at blokade af IKs også kunne bidrage til lægemiddelinduceret alkt ‘ er, specielt når repolariseringsreserven er kompromitteret (81). Det blev påvist, at IKS-egenskaberne undertrykkes, både in vivo og in vitro, og at de førte til markante grænseværdier (81). Polymorfismer, D85N i mink (gen KCNE1), T8A, K9E i MiRP1 (gen KCNE2), som er formodede larr-underenheder af IKs-og IKr-kanalerne, blev rapporteret at forårsage alkter (82, 83). Der er også rapporteret om Autoimmune antistoffer hos en patient med IgG indeholdende anti-HERG antistoffer (84).
elektrokardiografiske træk i de tre almindelige former for langt KVT-syndrom
typiske ST-T-bølgemønstre er til stede hos de fleste genotypede patienter og kan bruges til at identificere LKT1, LKT2 og muligvis LKT3 genotyper (85, 86).
LKT1-formen af LKT ‘ erne er forbundet med en bred T-bølge uden forkortelse af KVT-intervallet ved træning (figur 2a). 2 er forbundet med lav amplitude, ofte bifid, T bølger (figur 2b). 3 er forbundet med et langt iso-elektrisk segment og en smal-baseret, høj T-bølge (figur 2C). Pauseafhængigheden af TDP-indtræden i medfødte misdannelser er genotypespecifik, idet den er fremherskende i symptomer 2, men næsten fraværende i symptomer 1 (87).

figur 2. Elektrokardiogramoptagelser fra patienter med LKT1, LKT2 og LKT3. (A) 12-bly EKG af en 18-årig mand med en kcnk1-mutation. Dette interval er forlænget (KTC = 500 ms). ST-segmentet har en bred base og relativt stor amplitude. Ledningsintervallet er normalt (standardkalibrering). (B) 12-bly EKG af en 14-årig pige med en kcnh2-mutation. KVT-intervallet forlænges (Kvtc-520 ms). ST-segmentet er hakket i bly V3 og har relativt lav amplitude i ekstremitetsledningerne. Ledningsintervallet er normalt (standardkalibrering). (C) 12-bly EKG af en 12-årig dreng med en SCN5A-mutation. Dette interval er forlænget (600 ms). ST-segmentet har et langt (næsten) iso-elektrisk segment med en stor, skarp og smal T-bølge. Ledningsintervallet er normalt (standardkalibrering).
selvom mønstre kan tyde på en specifik genotype, er der ofte fundet undtagelser.
genotype-fænotype
langt KVT-syndrom er en autosomal dominerende sygdom. Genotype-og fænotypeanalyse blandt de heterosygøse mutationsbærere er blevet udført ret udførligt hos patienterne i KK1, KK2 og KK3. I barndommen er risikoen for hjertehændelser signifikant højere hos mænd i MKVTS1 end hos kvinder i MKVTS1, mens der ikke blev observeret signifikante kønsrelaterede forskelle i risikoen for hjertehændelser blandt patienter i MKVTS2 og MKVTS3 (88-91). I voksenalderen (også efter 40-årsalderen) kunne kvinder i 1 KKS og 2 KKS have signifikant højere risiko for hjertehændelser end respektive mænd (88-91). Generelt synes dødeligheden af hjertehændelser at være fremherskende hos LKTS3-patienter end hos LKTS1-og LKTS2-patienter (91). Kvinder med hjertelidelser har en reduceret risiko for hjertehændelser under graviditeten, men risikoen stiger ganske i løbet af 9-måneders postpartumperioden, specielt hos kvinder med mutation i KCNH2-genet (92).
pludselig hjertedød hos børn kan også være forårsaget af mutationer i hjerteionkanalgenerne (93-96). Cirka 28% af børnene med en uforklarlig SCD (mellem 1 og 18 år, gennemsnitsalder: 12,3 3,8 år) var bærere af mutationer i kausale gener (97). I SIDS forekom mutationer i SCN5A overvejende (98, 99), men mutationer i KCNK1, KCNH2, KCNE2 og CAV3, SCN4B og SCN3B blev også fundet (100, 101). Intrauterin føtal dødsfald blev også rapporteret på grund af defekter i hjerte-ionkanalgener (12, 102).
klinisk behandling af hjertelidelser
ophør af alle lægemidler, der vides at forlænge HJERTELIDELSESINTERVALLET, og også korrektion af elektrolytubalancer og/eller udfældende metaboliske tilstande bør være det primære fokus under behandling af (erhvervede) hjertelidelser. Derfor anbefales generelt begrænsning af patienters deltagelse i atletiske aktiviteter (24, 25). Grundpillerne i klinisk terapi for LKT ‘ erne er LKR-blokade. Langtidsvirkende præparater af propranolol, nadolol og metoprolol anvendes sædvanligvis, og deres effektivitet ved kurr-blokade vurderes ved at udøve hjertefrekvensen (f. eks. ved >20%) (24, 25). Blandt alle de karrus-blokkere, propranolol og nadolol anses overlegen end metoprolol hos symptomatiske patienter (103). Desuden kan Kurt-blokade også anvendes som en profylaktisk behandling i tavse mutationsbærere for at reducere SCD (24). I en undersøgelse foretaget af Barsheshet et al. (30) patienter med C-loop missense-mutationer i kcnk1-genet udviste høj risiko for livstruende hjertehændelser, og de havde betydelige fordele ved behandlingen med Kurt-blokkere. Da kvinder med HC2 har en øget risiko i løbet af 9-måneders postpartumperioden, bør der ordineres karrus-blokkere for at reducere eventuelle hjertehændelser i denne højrisikoperiode (92). En implanterbar cardioverter-defibrillator (ICD ‘ er) kan overvejes til patienter med tilbagevendende synkope på trods af behandling med en behandling med kronisk blokering eller hos patienter med høj risiko for hjertestop (f. eks., symptomatisk LKTS2 og LKTS3 med en dokumenteret forlængelse af Kks2). Venstre hjertesympatisk denervering (LCSD) anbefales til patienter med høj risiko, hvor en ICD er kontraindiceret eller afvist, og kursblokkere enten ikke er effektive, ikke tolereres, ikke accepteres eller kontraindiceret (18, 104). Jlns-patienter har normalt en KTC > 500 ms og er også i høj risiko, og Kristian-blokkere har utilstrækkelig effekt hos sådanne patienter, hvor en tidlig behandling med ICD blev anbefalet (18, 31).
: Saudi Perspective
den første rapport fra Saudi-Arabien blev offentliggjort i 1993 fra Riyadh Armed Forces Hospital (105). Fire spædbørn og små børn mellem 6 og 48 måneder med historie med tilbagevendende anfald, fra en enkelt familie, blev diagnosticeret som LKS (105). Familiehistorie afslørede, at to andre udvidede familiemedlemmer havde lignende episoder med pludseligt tab af bevidsthed, og tre familiemedlemmer døde pludselig (105). I alle tilfælde var den første diagnose epilepsi (105). Flere år senere er der rapporteret om to sporadiske sagsrapporter med relativt severere variant af neonatale LKT ‘ er kombineret med 2:1 AV-blok (106, 107). Alle disse offentliggjorte kliniske rapporter var uden genetiske fund, der kunne forklare patofysiologien af LKD ‘ er i dem (105-107).
vi har for første gang rapporteret genetiske defekter som et patologisk grundlag for LKS hos lignende patienter fra Saudi-Arabien. Vi har undersøgt seks saudiske familier med historie med synkope og pludselige uforklarlige dødsfald hos Foster, nyfødte og børn (12-14). Autosomal recessiv LKT1 blev diagnosticeret hos børn fra to familier (figur 3). Autosomal recessiv LKT2 blev diagnosticeret i to familier (figur 4). I en familie blev en kvindelig patient diagnosticeret med autosomal dominant LKTS2 (figur 5), patienten havde synkopale angreb under postpartumperioden på hospitalet, hvilket er meget almindeligt hos kcnh2-mutationsbærerkvinder. Hos alle vores patienter, identifikation af patogene mutationer i kausale hjerte-ionkanalgener førte til en bekræftet klinisk diagnose, som blev fejldiagnosticeret som epileptiske kramper, før de blev henvist til os (13, 14) for nylig, Shinvari et al. (15) fra King Faisal Specialist Hospital og Forskningscenter rapporterede en kausal kcnk1-mutation, H258P, i en stor familie med 12 berørte personer. Kun to bærere var symptomatiske, deres KTC var >500 ms, og kur-blokkere undertrykte de kliniske symptomer hos en patient, og den anden symptomatiske patient krævede en ICD.
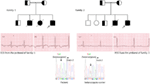
figur 3. Stamtavle tegning af familien-1 og 2 med autosomal recessiv LKT1, probands er vist med en pil. EKG ‘ er fra probands i to familier vises i midten med en Kvtc på henholdsvis 557 og 529 ms. 387 -5 t > A i KCNK1-genet blev fundet hos patienterne (vist i bunden). Mutation blev vist med en pil. Ikke-fyldte cirkler og firkanter er ikke-bærere af mutationen. Berørte personer vises som fyldte cirkler (kvindelige) og firkanter (mandlige). Halvfyldte firkanter og cirkler er individer med heterosygotmutation. Afdøde individer er angivet med skråstreger, probander er angivet med en pil, og sammenhængende ægteskab er angivet med = Ekson − intron grænse er vist med en stiplet linje med pil, der peger mod ekson.
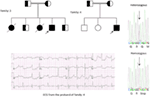
figur 4. Stamtavle tegning af familien-3 og 4 med autosomal recessiv REFERENCE2. EKG proband fra familie-4 er vist under stamtavlen tegning, som viser sinus takykardi, næsten komplet AV-blok, bred kompleks flugt rytme med meget lange KTC intervaller (KT >600 ms). Mutation, c. 3208 C > T (s.1070 gange) i kcnh2-genet blev fundet hos patienterne (vist til højre), markeret med en pil.
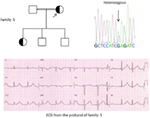
figur 5. Øverst til venstre: stamtavle af familien-5. Berørt person er vist med fyldt cirkel (Kvindelig) Proband er angivet med en pil. 12-bly EKG af proband (i:2). EKG viser en høj toppet, bred baseret, stor amplitude T-bølge, Kvtc-interval er 580 ms. Screening af KCNH2-genet viser substitution af nukleotid “G” for en ” A ” (c.2362g > a, pil markeret), hvilket fører til aminosyresubstitution, s. E788K.
genetiske og kliniske fund i Vores undersøgelse (12-14) fra Saudi-Arabien er ret spændende af flere grunde: (1) i alt har vi undersøgt seks familier, blandt dem fire var homosygøse / sammensatte heterosygøse for mutationerne, og mutationerne stammer fra en forfædres kilde; (2) alle mutationerne var nye, kun rapporteret i disse Arabiske familier; (3) på grund af homosygositeten eller den sammensatte heterosygositet for mutationerne var kliniske fænotyper også alvorlige i vores undersøgte familier (12-14). Vi foreslog, at de genetiske og fænotypiske observationer stammede fra den ekstreme høje grad af sammenhængende ægteskaber i Saudi-Arabien (108, 109). Vores undersøgelse gav det første videnskabelige bevis for konsanguinitetens rolle i at udøve en central rolle i uforklarlige hjertearytmier og SCDs hos børn og unge i Saudi-Arabien (12-14). Vi har også vist, at kcnk1-mutationen c.387 -5 t > A (NM_000218) (figur 3) havde spredt sig i Assir-provinsen Saudi-Arabien fra en fælles forfader i flere generationer på grund af høj forekomst af sammenhængende ægteskaber . Indtil videre er dette den mest almindelige kausale mutation i den saudiarabiske befolkning, som også blev observeret på King Faisal Specialist Hospital og på Khamis Mashayt Military Hospital (upubliceret). Et lignende resultat blev også opnået for kausal mutation C. 3208 C > T (s.4) i kcnh2-genet (12, 14), som også er en grundlæggermutation i Saudi-Arabien og adskilt i mange generationer i Assir-regionen (se kortet, figur 6). Vi ville forudsige, at der findes et betydeligt antal individer med de nævnte forfædres/grundlæggermutationer (både i KCNK1 og KCNH2 og andre gener) i denne region og også i de store byer som Riyadh, Jeddah og Dammam på grund af bymigration. Flere grundlægger-eller forfædres mutationer, der er patogene for LKT ‘ er, er meget planlagt i andre provinser i Saudi-Arabien på grund af høj grad af sammenhængende ægteskaber.
figur 6. Kort over Saudi-Arabien (med tilladelse). Assir-regionen er markeret med tyk linje, hvor vi har fundet grundlæggermutationerne i kcnk1-og KCNH2-gener, beskrevet i familie 1-4.
da det er en autosomal dominant sygdom, forventede vi at observere overvejende heterosygøse mutationsbærerpatienter. Men i vores undersøgelse identificerede vi hovedsageligt patienter med recessive mutationer (12-14). Klart, disse patienter bliver først symptomatiske og, blev overvejende henvist til Prince Sultan Cardiac Center, bringe dem under vores opmærksomhed. Ikke desto mindre indebærer resultater fra vores undersøgelser det faktum, at recessive kliniske fænotyper kan have fatale kliniske fænotyper hos børn, og de er ikke ualmindelige i Saudi-Arabien (12-14). Da de heterosygøse mutationsbærere også er modtagelige for at udvikle arytmi og dens komplikationer, bør der tages samordnet initiativ for at bringe de lokale generelle læger, kardiologer og kliniske genetikere på en fælles platform for at identificere de personer, der er i fare. Desuden har vi i øjeblikket heller ingen genetisk information for andre familiære arytmiforstyrrelser, f.eks. Specialiserede kardiogenetiske Centre bør tage initiativ til at søge efter de genetiske defekter, mutationer og udføre genotype-fænotypestudier i alle former for arvelige arytmier. Det skal også tages i betragtning, at ikke alle genetiske arytmier ville have en familiehistorie, da arytmi-kausal mutation i mange tilfælde er de novo-oprindelse, dvs.Probanden er den første patient i den familie med mutationen, og han/hun er kilden til at overføre mutationen i efterfølgende generationer (110, 111). På grund af den meget høje grad af sammenhængende ægteskaber i Saudi-Arabien, vi forventer, at meget flere grundlæggermutationer udøver en afgørende rolle i medfødte arytmier i dette land. At identificere disse grundlæggermutationer bør være vores først og fremmest opgave, hvilket ville lette os med at udvikle en effektiv førægteskabelig og også præsymptomatisk genetisk rådgivning i dette land. I en ekstrem, nogle bærere kunne formodentlig være helt sunde, men nogle bærere kunne have deres første manifestation af sygdommen som synkope eller pludselig død. Pludselig død af et lille barn eller voksen udgør en stor psykologisk og følelsesmæssig byrde for familien, screening af transportørerne er afgørende, da der findes simpel medicin, f.eks. Personer med homogene mutationer i kausale gener kan have alvorlig sygelighed og ekstrem høj dødelighed inklusive føtal brady-takyarytmier, og i mange tilfælde aborter, spontane aborter hos gravide mødre (12-14).
der er også udført ikke meget forskning i Saudi-Arabien om forekomsten af forskellige SNP ‘ er i de arytmiforbundne gener og også i de gener, der regulerer dem. Et al. (112) beskrev en almindelig s1103y-variant i SCN5A-genet forbundet med arytmi hos afroamerikanere. Varianten allel betegnet Y1103 er ansvarlig for at accelerere kanalaktivering og derved øge sandsynligheden for hjertearytmier hos personer af afrikansk afstamning (52, 112). K393N er en variant i kcnk1 gen rapporteret hos LKTS1 patienter i USA, men i den arabiske befolkning har vi påvist denne variant hos 2% af individer (upublicerede data). Hvorvidt k393n-varianten i Kcnk1-genet hos arabere er sammenlignelig med s1103y (SCN5A)-varianten hos afroamerikanere eller d85n (KCNE1) – varianten i den japanske befolkning kunne også fortjene undersøgelse (59).
interessekonflikt Erklæring
forfatterne erklærer, at forskningen blev udført i mangel af kommercielle eller økonomiske forhold, der kunne fortolkes som en potentiel interessekonflikt.
anerkendelser
vores oprigtige taknemmelighed til professor Arthur, Professor Connie Besina og udgiveren af tidsskriftet “Heart” for deres tilladelse til at bruge Figur 1 i dette manuskript fra Ref. (113).
1. Bhuiyan til. Klinisk og genetisk spektrum af arvelige hjertearytmi syndromer. Amsterdam: Amsterdam Universitet (2009).
2. Priori SG, Aliot E, bl Strom-Lundvistc, Bossaert L, Breithardt G, Brugada P, et al. Task force om pludselig hjertedød, European Society of cardiology. Europace (2002) 4: 3-18. doi: 10.1053 / eupc.2001.0214
CrossRef Fuld Tekst
3. I, Atkinson D, Li, Robinson JL, et al. SCN5A-mutationer forbundet med en arvelig hjertearytmi, langt KT-syndrom. Celle (1995) 80:805-11. doi:10.1016/0092-8674(95)90359-3
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
4. Curran mig, Splash I, Timothy KVA, Vincent GM, grøn ED, Keating MT. Et molekylært grundlag for hjertearytmi: HERG-mutationer forårsager langt KT-syndrom. Celle (1995) 80:795-803. doi: 10.1016/0092-8674(95)90358-5
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
5. Jeg er en af de mest populære i verden, og jeg er en af de bedste i verden. Positionel kloning af et nyt kaliumkanalgen: kvlkt1-mutationer forårsager hjertearytmier. Nat Genet (1996) 12:17-23. doi:10.1038 / ng0196-17
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
6. Lehmann MH, Sanguinetti MC, Keating MT. Mutationer i hminK-genet forårsager langt KT-syndrom og undertrykker IKs-funktion. Nat Genet (1997) 17:338-40. doi:10.1038 / ng1197-338
Pubmed abstrakt | Pubmed Fuld tekst | CrossRef Fuld tekst
7. Sanguinetti MC, Jiang C, Curran mig, Keating MT. En mekanistisk forbindelse mellem en arvelig og en erhvervet hjertearytmi: HERG koder for IKr-kaliumkanalen. Celle (1995) 81:299-307. doi:10.1016/0092-8674(95)90340-2
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. En ny mutation i kaliumkanalgenet KVLKT1 forårsager jervell og lange-Nielsen cardioauditory syndrom. Nat Genet (1997) 15:186-9. doi:10.1038 / ng0297-186
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
9. Kucera JP, Rohr S, Rudy Y. lokalisering af natriumkanaler i interkalerede diske modulerer hjerteledning. Circ Res (2002) 91:1176-82. doi: 10.1161 / 01.RES.0000046237.54156. 0 a
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
10. Maass K, Coombs V, Taffet SM, Delmar M. Forbindelsein43 remodeling forårsaget af inhibering af plakophilin-2 ekspression i hjerteceller. Circ Res (2007) 101: 703-11. doi: 10.1161 / CIRCRESAHA.107.154252
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
11. Rohr S. Molekylær krydstale mellem mekaniske og elektriske kryds ved den interkalerede skive. Circ Res (2007) 101: 637-9. doi: 10.1161 / CIRCRESAHA.107.161901
CrossRef Fuld Tekst
12. Bhuiyan til, Momenah TS, Gong K, Amin AS, Ghamdi SA, Carvalho JS, et al. Tilbagevendende intrauterin føtal tab på grund af næsten fravær af HERG: klinisk og funktionel karakterisering af en homosygøs nonsens HERG 1070h-mutation. Hjerterytme (2008) 5:553-61. doi: 10.1016 / j. hrthm.2008.01.020
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
13. Bhuiyan til, Momenah TS, Amin TS, Al-Khadra AS, Alders M, vil AAM, et al. En Intronisk mutation, der fører til ufuldstændig springning af ekson-2 i KCNK1, redder hørelsen i Jervell og Lange-Nielsen syndrom. Prog Biophys Mol Biol (2008) 98:319-27. doi: 10.1016 / j. pbiomolbio.2008.10.004
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
14. Bhuiyan til, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi S, Al-Kalaf K, Mannens MMAM, et al. Klinisk og genetisk analyse af langt KT-syndrom hos børn fra seks familier i Saudi-Arabien: er de forskellige? Pediatr Cardiol (2009) 30:490-501. doi: 10.1007 / s00246-008-9377-y
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
15. Det er et af de mest populære områder i verden. Identifikation af en ny kcnk1-mutation i en stor saudisk familie med langt KT-syndrom: kliniske konsekvenser og forebyggende implikationer. Clin Genet (2013) 83:370-4. doi: 10.1111 / j. 1399-0004.2012. 01914.
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
16. Pj, Stramba-Badiale M, Crotti L, Pedrasini M, Besana a, Bosi G, et al. Prævalens af det medfødte lang-KT syndrom. Cirkulation (2009) 120: 1761-7. doi:10.1161 / CIRCULATIONAHA.109.863209
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
17. PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostiske kriterier for det lange KT-syndrom. Opdatering. Cirkulation (1993) 88:782-4. doi: 10.1161 / 01.CIR.88.2.782
CrossRef Fuld Tekst
18. Priori SG, vil AA, Horie M, Cho Y, Behr ER, Berul C, et al. Sammendrag: hrs/EHRA / APHRS konsensuserklæring om diagnose og behandling af patienter med arvelige primære arytmisyndromer. Europace (2013) 15: 1389-406. doi: 10.1093 / europace / eut272
CrossRef fuldtekst
19. Liu JF, Jons C, Moss AJ, McNitt s, Peterson DR, hvem, et al. Syndrom Register. Risikofaktorer for tilbagevendende synkope og efterfølgende fatale eller næsten fatale hændelser hos børn og unge med langt KT-syndrom. J Am Coll Cardiol (2011) 57:941-50. doi: 10.1016 / j. jacc.2010.10.025
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
20. I, Shen J, Timothy KV, Lehmann MH, Priori S, Robinson JL, et al. Spektrum af mutationer i lang-KT syndrom gener. KVLKV1, HERG, SCN5A, KCNE1 og KCNE2. Cirkulation (2000) 102: 1178-85. doi: 10.1161 / 01.CIR.102.10.1178
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
21. I, Timothy K, Keating MT, Sanguinetti MC. Sammensatte mutationer: en almindelig årsag til svær lang-KT syndrom. Cirkulation (2004) 109: 1834-41. doi: 10.1161 / 01.CIR.0000125524.34234.13
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
22. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Sareba M, et al. Risiko for livstruende hjertehændelser blandt patienter med langt KT-syndrom og multiple mutationer. Hjerterytme (2013) 10: 378-82. doi: 10.1016 / j. hrthm.2012.11.006
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
23. PJ, Bloise R, Ronchetti E, Nastoli J, et al. Udvikling og validering af en effektiv tilgang til genotypebestemmelse i klinisk praksis. JAMA (2005) 294: 2975-80. doi: 10.1001 / jama.294.23.2975
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
24. Roden DM. Klinisk praksis. Lang-KT syndrom. N Engl J Med (2008) 358:169-76. doi: 10.1056 / NEJMcp0706513
CrossRef Fuld tekst
25. PJ, Priori SG, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-fænotype-korrelation i det lange KT-syndrom: genspecifikke udløsere for livstruende arytmier. Cirkulation (2001) 103: 89-95. doi: 10.1161 / 01.CIR.103.1.89
CrossRef Fuld Tekst
26. Ackerman MJ, Tester DJ, Porter CJ. Svømning, en genspecifik arytmogen trigger til nedarvet langt KT-syndrom. Mayo Clin Proc (1999) 74:1088-94. doi:10.4065/74.11.1088
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
27. Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, et al. Genetisk testning for lang-KT syndrom: skelne patogene mutationer fra godartede varianter. Cirkulation (2009) 120: 1752-60. doi:10.1161 / CIRCULATIONAHA.109.863076
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
28. Moss AJ, Shimusu m, ville AA, anhængertræk JA, Sareba m, Robinson JL, et al. Kliniske aspekter af type 1-syndrom efter placering, kodende type og biofysisk funktion af mutationer, der involverer kcnk1-genet. Cirkulation (2007) 115: 2481-9. doi:10.1161 / CIRCULATIONAHA.106.665406
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
29. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. Varianter i det 3 ‘ uoversatte område af den KCNK1-kodede Kv7.1 kaliumkanal modificerer sygdommens sværhedsgrad hos patienter med type 1 Langt KT-syndrom på en allelspecifik måde. Eur Hjerte J (2012) 33: 714-23. doi: 10.1093 / eurheartj / ehr473
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
30. Barsheshet A, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimisu M, et al. Mutationer i cytoplasmatiske sløjfer i kcnk1-kanalen og risikoen for livstruende hændelser: implikationer for mutationsspecifik respons på behandling med Kurt-blokker i type 1-syndrom. Cirkulation (2012) 125: 1988-96. doi:10.1161 / CIRCULATIONAHA.111.048041
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
31. C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. Jervell og lange-Nielsen syndromet: naturhistorie, molekylær basis og klinisk resultat. Cirkulation (2006) 113: 783-90. doi:10.1161 / CIRCULATIONAHA.105.592899
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
32. Bhuiyan til, Vil AA. Ik i hjerte og hørelse, øret kan gøre med mindre end hjertet. Circ Cardiovasc Genet (2013) 6:141-3. doi:10.1161 / CIRCGENETICS.113.000143
CrossRef Fuld Tekst
33. B, Schroeder f. kr., Kubisch C, Esperer HD, Jentsch TJ. Patofysiologiske mekanismer for dominerende og recessive kvlkt1 k + kanal mutationer fundet i arvelige hjertearytmier. Hum Mol Genet (1997) 6:1943-9. doi: 10.1093 / hmg / 6.11.1943
CrossRef Fuld Tekst
34. Han er en af de mest populære i verden, og han er en af de bedste i verden. Auditiv stimuli som udløser for arytmiske hændelser differentierer HERG-relaterede (LKT2) patienter fra KVKT1-relaterede patienter (LKT1). J Am Coll Cardiol (1999) 33:327-32. doi: 10.1016 / S0735-1097(98)00578-6
CrossRef Fuld tekst
35. Moss AJ, Ba, Kaufman ES, Gartman E, Peterson DR, Benhorin J, et al. Med mutationer i poreområdet af den humane ether-a-go-go-relaterede genkaliumkanal. Cirkulation (2002) 105: 794-9. doi:10.1161/hc0702.105124
Pubmed abstrakt | Pubmed Fuld tekst | CrossRef Fuld tekst
36. Han er en af de mest kendte i verden, og han er en af de største i verden. Lange KT syndrom hos nyfødte: ledningsforstyrrelser forbundet med HERG mutationer og sinus bradykardi med kcnk1 mutationer. J Am Coll Cardiol (2004) 43:826-30. doi: 10.1016 / j. jacc.2003.09.049
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
37. P, P, M, M, P, Potetf, Schott JJ, et al. Torsades de pointes komplicerer atrioventrikulær blok: bevis for en genetisk disponering. Hjerterytme (2007) 4: 170-4. doi: 10.1016 / j. hrthm.2006.10.004
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
38. Hoorntje T, Alders M, van Tintelen P, van der Lip K, Sreeram N, van der Val A, et al. Homosygøs for tidlig afkortning af HERG-proteinet: den humane HERG-knockout. Cirkulation (1999) 100: 1264-7. doi: 10.1161 / 01.CIR.100.12.1264
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
39. Piippo K, Laitinen P, Svane H, Toivonen L, Viitasalo M, Pasternack M, et al. En af de mest almindelige årsager til denne sygdom er, at det er en af de mest almindelige årsager til denne sygdom. J Am Coll Cardiol (2000) 35:1919-25. doi: 10.1016 / S0735-1097(00)00636-7
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
40. Johnson, der er Jr., Yang P, Yang T, Lau år, Mostella BA, Ulff DJ, et al. Det er en af de mest almindelige årsager til, at en person lider af denne sygdom. Pediatr Res (2003) 53:744-8. doi: 10.1203 / 01.PDR.0000059750.17002.B6
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
41. Ackerman MJ, Tester DJ, Jones GS, vil ML, grave CR, Curran mig. Etniske forskelle i hjertekaliumkanalvarianter: implikationer for genetisk modtagelighed for pludselig hjertedød og genetisk test for medfødt langt KT-syndrom. Mayo Clin Proc (2003) 78:1479-87. doi:10.4065/78.12.1479
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
42. C, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T er en genetisk modifikator af latent medfødt lang-KT syndrom. Cirkulation (2005) 112: 1251-8. doi:10.1161 / CIRCULATIONAHA.105.549071
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
43. Nof E, Cordeiro JM, P for at gøre det, Scornik FS, Calloe K, Kærlighed B, et al. En almindelig enkelt nukleotidpolymorfisme kan forværre type 2-syndrom, der fører til pludselig spædbarnsdød. Circ Cardiovasc Genet (2010) 3:199-206. doi:10.1161 / CIRCGENETICS.109.898569
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
44. Det er et af de mest populære områder i verden. En enkelt na ( + )-kanalmutation, der forårsager både lang-og Brugada-syndromer. Circ Res (1999) 85:1206-13. doi: 10.1161 / 01.RES.85.12.1206
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
45. Kyndt F, Probst V, Potetf, Demolombe S, Chevallier JC, Baro I, et al. Novel SCN5A-mutation, der fører enten til isoleret hjerteledningsdefekt eller Brugada-syndrom i en stor fransk familie. Cirkulation (2001) 104: 3081-6. doi:10.1161 / hc5001. 100834
Pubmed abstrakt | Pubmed Fuld tekst | CrossRef Fuld tekst
46. Makita N, Behr E, Shimisu m, Horie M, Sunami A, Crotti L, et al. E1784k-mutationen i SCN5A er forbundet med blandet klinisk fænotype af type 3 langt KVT-syndrom. J Clin Invest (2008) 118: 2219-29. doi:10.1172 / JCI34057
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
47. Keller DI, Acharfi S, Delacr, Benammar N, Rotter M, Pfammatter JP, et al. En ny mutation i SCN5A, delkkp 1507-1509, der forårsager langt KVT-syndrom: rolle af 1507-Rest i natriumkanalinaktivering. J Mol Celle Cardiol (2003) 35:1513-21. doi: 10.1016 / j. yjmcc.2003.08.007
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
48. PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risikostratificering i det lange KT-syndrom. N Engl J Med (2003) 348:1866-74. doi:10.1056 / NEJMoa022147
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
49. Jeg er en af dem, der er interesseret i at finde ud af, hvad der er bedst. Homosygøs scn5a-mutation i lang KT-syndrom med funktionel to-til-en atrioventrikulær blok. Circ Res (2001) 89:E16–21. doi: 10.1161 / Hh1401. 095087
CrossRef Fuld tekst
50. Shi R, yang y, Yang C, Huang C, Yang H, et al. Hjertenatriumkanalmutationen delkkp 1507-1509 er forbundet med det ekspanderende fænotypiske spektrum af LKT3, ledningsforstyrrelse, dilateret kardiomyopati og høj forekomst af ungdoms pludselige død. Europace (2008) 10: 1329-35. doi: 10.1093 / europace / eun202
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
51. B, Baartscheer A, Eckardt L, Paul M, et al. En heterosygøs deletionsmutation i hjertenatriumkanalgenet SCN5A med Tabs – og forstærkningsfunktionskarakteristika manifesterer sig som isoleret ledningssygdom uden tegn på Brugada eller langt KT-syndrom. PLoS One (2013) 8: e67963. doi:10.1371 / tidsskrift.pone.0067963
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
52. Plant LD, bueskytter PN, Liu P, Morgan T, Jang T, stat mv, Et Al. En almindelig hjertenatriumkanalvariant forbundet med pludselig spædbarnsdød hos afroamerikanere, SCN5A S1103Y. J Clin Invest (2006) 116:430-5. doi: 10.1172 / JCI25618
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
53. Mohler PJ, Schott JJ, Gramolini AO, Dilly KV, Guatimosim S, duBell et al. Ankyrin-B-mutation forårsager hjertearytmi af type 4 og pludselig hjertedød. Natur (2003) 421: 634-9. doi:10.1038 / nature01335
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
54. Schott JJ, Charpentier F, Peltier s, Foley P, Drouin E, Bouhour JB, et al. Kortlægning af et gen for langt KT-syndrom til kromosom 4k25-27. Am J Hum Genet (1995) 57:1114-22.
Pubmed Abstrakt / Pubmed Fuld Tekst
55. Barhanin J, Lesage F, Guillemare E, Fink M, Lasdunski M, Romey G. K(V)LKT1 og LSK (minK) proteiner associerer til dannelse af i(Ks) hjertekaliumstrøm. Natur (1996) 384: 78-80. doi:10.1038 / 384078a0
Pubmed abstrakt | Pubmed Fuld tekst | CrossRef Fuld tekst
56. Sanguinetti MC, Curran mig, til en, Shen J, Spector PS, Atkinson DL, et al. Coassembly af K (V)LKT1 og minK(isk) proteiner til dannelse af hjerte-i (Ks) kaliumkanal. Natur (1996) 384:80-3. doi: 10.1038 / 384080a0
Pubmed abstrakt | Pubmed Fuld tekst / CrossRef Fuld tekst
57. Det er et af de mest populære områder i verden. Kcne1-mutationer forårsager jervell og Lange-Nielsen-syndrom. Nat Genet (1997) 17:267-8. doi: 10.1038 / ng1197-267
CrossRef Fuld tekst
58. D, Villafane J, Kaushik V, Beggs AH. Mutation af genet for IKs forbundet med både Jervell og Lange-Nielsen og Romano-afdeling former for lang-KT syndrom. Cirkulation (1998) 97:142-6. doi: 10.1186/1471-2350-9-24
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YS, et al. D85n, en kcne1-polymorfisme, er en sygdomsfremkaldende genvariant i langt KT-syndrom. J Am Coll Cardiol (2009) 54:812-9. doi: 10.1016 / j. jacc.2009.06.005
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
60. Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, et al. Genetiske variationer af KCNK1, KCNH2, SCN5A, KCNE1 og KCNE2 hos patienter med lægemiddelinduceret langt KT-syndrom. Mol Med (2004) 82:182-8. doi: 10.1007 / s00109-003-0522-
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
61. Jeg er en af dem, der er interesseret i at finde ud af, hvad der er bedst. MiRP1 danner IKr-kaliumkanaler med HERG og er forbundet med hjertearytmi. Celle (1999) 97:175-87. doi:10.1016 / S0092-8674 (00) 80728 –
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
62. Gordon E, Panaghie G, Deng L, Bee KJ, Roepke TK, Krogh-Madsen T, et al. En kcne2-mutation hos en patient med hjertearytmi induceret af auditiv stimuli og serumelektrolytubalance. Cardiovasc Res (2008) 77:98-106. doi:10.1093 | CVR | cvm030
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
63. Gips NM, vil R, Tristani-Firous M, kan være S, Bendahhou S, Tsunoda A, et al. Mutationer i Kir2.1 forårsager udviklingsmæssige og episodiske elektriske fænotyper af Andersens syndrom. Celle (2001) 105: 511-9. doi: 10.1016 / S0092-8674(01)00342-7
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
64. Kubo Y, Balduin TJ, Jan YN, Jan LY. Primær struktur og funktionel ekspression af en mus indad ensretter kalium kanal. Natur (1993) 362:127-33. doi:10.1038 / 362127a0
Pubmed abstrakt | Pubmed Fuld tekst | CrossRef Fuld tekst
65. Raab-Graham KF, Radeke CM, Vandenberg CA. Molekylær kloning og ekspression af et menneskeligt hjerte indad ensretter kalium kanal. Neuroreport (1994) 5:2501-5. doi: 10.1097/00001756-199412000-00024
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
66. I, Timothy, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca (V) 1.2 calciumkanaldysfunktion forårsager en multisystemforstyrrelse, herunder arytmi og autisme. Celle (2004) 119: 19-31. doi: 10.1016 / j.celle.2004.09.011
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
67. I, Timothy, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Alvorlig arytmiforstyrrelse forårsaget af hjerte-L-type calciumkanalmutationer. Proc Natl Acad Sci U S A (2005) 102:8089-96. doi: 10.1073 / pnas.0502506102
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
68. Gillis J, Burashnikov E, Antselevitch C, Blaser S, Gross G, Turner L, et al. Lang, syndaktisk, ledkontrakturer, slagtilfælde og ny cacna1c-mutation: udvidelse af spektret af Timothy syndrom. Am J med Genet A (2012) 158A: 182-7. doi: 10.1002 / ajmg.a.34355
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
69. Dufendach KA, Giudicessi JR, Bocek NJ, Ackerman MJ. Maternal mosaicisme forvirrer den neonatale diagnose af type 1 Timothy syndrom. Pædiatri (2013) 131: e1991–5. doi: 10.1542 / peds.2012-2941
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
70. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanse EE, Taylor et al. Mutant caveolin – 3 inducerer vedvarende sen natriumstrøm og er forbundet med lang-KT syndrom. Cirkulationen (2006) 114:2104–12. doi:10.1161 / CIRCULATIONAHA.106.635268
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
71. Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, et al. Ny mekanisme til pludseligt spædbarnsdødssyndrom: vedvarende sen natriumstrøm sekundært til mutationer i caveolin-3. Hjerterytme (2007) 4: 161-6. doi: 10.1016 / j. hrthm.2006.11.030
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
72. Engelman JA, Galbiati F, Volonte D, Sotgia F, Pestell RG, et al. Molekylær genetik af caveolin-genfamilien: implikationer for humane kræftformer, diabetes, sygdom og muskeldystrofi. Am J Hum Genet (1998) 63:1578-87. doi:10.1086/302172
CrossRef Fuld tekst
73. Balijepalli RC, Foell JD, Hall DD, helvede JV, Kamp TJ. Lokalisering af hjerte-L-Type Ca(2+) kanaler til et caveolært makromolekylært signalkompleks er påkrævet til beta (2)-adrenerg regulering. Proc Natl Acad Sci U S A (2006) 103:7500-5. doi: 10.1073 / pnas.0503465103
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
74. Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty a, Ye B, et al. SCN4B-kodet natriumkanalbeta4-underenhed i medfødt lang-KT-syndrom. Cirkulation (2007) 116: 134-42. doi:10.1161 / CIRCULATIONAHA.106.659086
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
75. Chen L, Markardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation af et a-kinase-forankringsprotein forårsager lang-KT syndrom. Proc Natl Acad Sci U S A (2007) 104:20990-5. doi: 10.1073 / pnas.0710527105
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
76. JJ, Mohapatra B, JJ, et al. Alfa-1-syntrofinmutation og det lange KT-syndrom: en sygdom med natriumkanalforstyrrelser. Circ Arrytme Electrophysiol (2008) 1: 193-201. doi: 10.1161 / CIRCEP.108.769224
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, et al. Identifikation af en Kir3.4 mutation i medfødt langt KT-syndrom. Am J Hum Genet (2010) 86:872-80. doi: 10.1016 / j.ajhg.2010.04.017
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
78. Roden DM. Mekanismer og styring af proarytmi. Am J Cardiol (1998) 82:49I–57I. doi:10.1016/S0002-9149(98)00472-
CrossRef Fuld tekst
79. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. Et strukturelt grundlag for lægemiddelinduceret langt KT-syndrom. Proc Natl Acad Sci U S A (2000) 97:12329-33. doi: 10.1073 / pnas.210244497
CrossRef Fuld Tekst
80. Kannankeril PJ, Roden DM. Lægemiddelinduceret lang tid og torsade de pointes: nylige fremskridt. Curr Opin Cardiol (2007) 22:39-43. doi: 10.1097 / HCO.0b013e32801129eb
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
81. Veerman CC, Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC, et al. Langsom forsinket ensretter kaliumstrømblokade bidrager vigtigt til lægemiddelinduceret langt KT-syndrom. Circ Arrytme Electrophysiol (2013) 6:1002-9. doi: 10.1161 / CIRCEP.113.000239
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
82. Seti F, Abbott G. J., Murray KT, Saksena S, PJ, et al. En almindelig polymorfisme forbundet med antibiotikainduceret hjertearytmi. Proc Natl Acad Sci U S A (2000) 97:10613-8. doi: 10.1073 / pnas.180223197
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
83. Det er en af de mest almindelige måder at gøre det på. En stor kandidatgenundersøgelse identificerer kcne1 d85n polymorfisme som en mulig modulator af lægemiddelinduceret torsades de pointes. Circ Cardiovasc Genet (2012) 5:91-9. doi:10.1161 / CIRCGENETICS.111.960930
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
84. Nakamura K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase s, et al. Anti-KCNH2-antistofinduceret langt KT-syndrom: ny erhvervet form for langt KT-syndrom. J Am Coll Cardiol (2007) 50:1808-9. doi: 10.1016 / j. jacc.2007.07.037
CrossRef Fuld Tekst
85. Moss AJ, Benhorin J, Locati EH, Hall J, Robinson JL, et al. EKG – t-bølgemønstre i genetisk forskellige former for det arvelige lange KT-syndrom. Cirkulation (1995) 92:2929-34. doi: 10.1161 / 01.CIR.92.10.2929
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
86. J, J, J, J, et al. Spektrum af ST-T-bølgemønstre og repolarisationsparametre i medfødt lang-KT syndrom: EKG-fund identificerer genotyper. Cirkulation (2000) 102: 2849-55. doi: 10.1161 / 01.CIR.102.23.2849
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
87. HL, Bardai A, Moss Aj, Schulse-Bahr E, Noda T, et al. Genotype-specifik begyndelse af arytmier ved medfødt lang-KT-syndrom: mulige terapiimplikationer. Cirkulation (2006) 114: 2096-103. doi:10.1161 / CIRCULATIONAHA.106.642694
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
88. Lehmann MH, et al. Alder-og kønsrelaterede forskelle i kliniske manifestationer hos patienter med medfødt langkvt-syndrom: fund fra Det Internationale KKT-Register. Cirkulation (1998) 97:2237-44. doi: 10.1161 / 01.CIR.97.22.2237
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
89. Jørgensen, Jørgensen, Jørgensen, Jørgensen, Jørgensen, Jørgensen, et al. Indflydelse af genotype på det kliniske forløb af lang-KT syndrom. International lang-KT syndrom Registry Research Group. N Engl J Med (1998) 339:960-5. doi: 10.1056 / NEJM199810013391404
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
90. Goldenberg I, Moss AJ, Bradley J, Polonsky s, Peterson DR, McNitt s, et al. Syndrom efter 40 år. Cirkulation (2008) 117: 2192-201. doi:10.1161 / CIRCULATIONAHA.107.729368
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
91. Lehmann MH, Peterson DR, Hall et al. Syndrom Register. Modulerende virkninger af alder og køn på det kliniske forløb af langt KT-syndrom efter genotype. J Am Coll Cardiol (2003) 42:103-9. doi: 10.1016 / S0735-1097(03)00554-0
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
92. Seth R, Moss AJ, McNitt S, S, M, M, M, M, et al. Lang tids syndrom og graviditet. J Am Coll Cardiol (2007) 49: 1092-8. doi: 10.1016 / j. jacc.2006.09.054
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
93. Pj, Priori SG, Dumaine R, Napolitano C, Antselevitch C, Stramba-Badiale M, et al. En molekylær forbindelse mellem det pludselige spædbarnsdødssyndrom og det lange KT-syndrom. N Engl J Med (2000) 343:262-7. doi: 10.1056 / NEJM200007273430405
CrossRef Fuld tekst
94. Pj, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, et al. Molekylær diagnose hos et barn med pludseligt spædbarnsdødssyndrom. Lancet (2001) 358:1342-3. doi: 10.1016 / S0140-6736(01)06450-9
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
95. Christiansen M, T Krisner N, Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutationer i HERG K+-ion-kanalen: en ny forbindelse mellem langt KT-syndrom og pludseligt barnedødssyndrom. Am J Cardiol (2005) 95:433-4. doi: 10.1016 / j. amjcard.2004.09.054
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
96. Tester DJ, Ackerman MJ. Genetisk test for pludselig uforklarlig død hos de unge. J Am Coll Cardiol (2007) 49:240-6. doi: 10.1016 / j. jacc.2006.10.010
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
97. Hofman N, Tan HL, Clur SA, Alders M, van Langen IM, vil AA. Bidrag fra arvelig hjertesygdom til pludselig hjertedød i barndommen. Pædiatri (2007) 120: e967–73. doi: 10.1542 / peds.2006-3751
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
98. JP, Arnold R, Veldkamp MVH, Bajanovski T, et al. De novo-mutation i SCN5A-genet forbundet med tidlig begyndelse af pludselig spædbarnsdød. Cirkulation (2001) 104: 1158-64. doi:10.1161 / hc3501. 095361
CrossRef Fuld tekst
99. L, Arvestad M, Insolia R, Pedrasini M, et al. Hjertenatriumkanaldysfunktion ved pludseligt spædbarnsdødssyndrom. Cirkulation (2007) 115: 368-76. doi:10.1161 / CIRCULATIONAHA.106.646513
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
100. Van Norstrand, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molekylær og funktionel karakterisering af nye glycerol-3-phosphatdehydrogenase 1-lignende gen (GPD1-L) mutationer i pludseligt spædbarnsdødssyndrom. Cirkulation (2007) 116: 2253-9. doi: 10.1161 / CIRCULATIONAHA.107.704627
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
101. Tan BH, Pundi KN, Van Norstrand DV, Valdivia CR, Tester DJ, Medeiros-Domingo A, et al. Sudden infant Death syndrome-associerede mutationer i natriumkanalens beta-underenheder. Hjerterytme (2010) 7:771-8. doi: 10.1016 / j. hrthm.2010.01.032
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
102. Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, et al. Tilbagevendende fostertab i tredje trimester og maternel mosaicisme for langvarigt syndrom. Cirkulation (2004) 109: 3029-34. doi: 10.1161 / 01.CIR.0000130666.81539.9 E
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
103. Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Ikke alle betablokkere er ens i håndteringen af lang KT syndrom type 1 og 2: højere gentagelser af hændelser under Metoprolol. J Am Coll Cardiol (2012) 60:2092-6. doi: 10.1016 / j. jacc.2012.07.046
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
104. Pj, Priori SG, Cerrone M, Odero A, Napolitano C, et al. Venstre hjertesympatisk denervering i behandlingen af højrisikopatienter, der er ramt af lang-KT-syndromet. Cirkulation (2004) 109: 1826-33. doi: 10.1161 / 01.CIR.0000125523.14403.1 E
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
105. Singh B, Habbab MA, al Deeb SM, Biary N. idiopatisk langt KT-syndrom: at stille det rigtige spørgsmål. Lancet (1993) 341:741. doi:10.1016/0140-6736(93)90501-7
CrossRef Fuld tekst
106. Gorgels AP, Al Fadley F, tid L, Kantoch MJ, Al Halees A. Det lange KT-syndrom med nedsat atrioventrikulær ledning: en ondartet variant hos spædbørn. J Cardiovasc Electrophysiol (1998) 9:1225-32. doi: 10.1111 / j. 1540-8167.1998.tb00096.
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
107. Kantoch MJ, Korashi MM, Bulbul til, Gorgels AP. En nyfødt med en kompleks medfødt hjertesygdom, atrioventrikulær blok og torsade de pointes ventrikulær takykardi. Pacing Clin Electrophysiol (1998) 21: 2664-7. doi: 10.1111 / j. 1540-8159.1998.tb00043.
CrossRef Fuld tekst
108. Han er en af de mest kendte og mest kendte mennesker i verden. Konsanguinitet blandt den saudiarabiske befolkning. J Med Genet (1995) 32:623-6. doi: 10.1136 / jmg.32.8.623
CrossRef Fuld Tekst
109. Jeg elsker, Al Salloum AA, Al Herbish AS, Churachi MM, Al Omar AA. Konsanguinitet og større genetiske lidelser hos saudiske børn: en samfundsbaseret tværsnitsundersøgelse. Ann Saudi Med (2008) 28:169-73. doi:10.4103/0256-4947.51726
Pubmed abstrakt / Pubmed Fuld tekst / CrossRef Fuld tekst
110. Medeiros-Domingo A, Bhuiyan til, Tester DJ, Hofman N, Bikker H, van TINTELEN JP, et al. Den ryr2-kodede ryanodinreceptor / calciumfrigivelseskanal hos patienter, der tidligere er diagnosticeret med enten catecholaminerg polymorf ventrikulær takykardi eller genotype-negativt, træningsinduceret langt KVT-syndrom: en omfattende åben læseramme mutationsanalyse. J Am Coll Cardiol (2009) 54:2065-74. doi: 10.1016 / j. jacc.2009.08.022
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
111. Al-Aama JY, al-Ghamdi S, Bdier AY, vil AA, Bhuiyan til. De novo mutation i kcnk1-genet kausal til Jervell og Lange-Nielsen syndrom. Clin Genet (2013). doi: 10.1111 / cge.12300
Pubmed Abstrakt / Pubmed Fuld Tekst / CrossRef Fuld Tekst
112. Jeg, Timothy KV, Tateyama M, Clancy CE, Malhotra A, Beggs AH, et al. Variant af SCN5A natriumkanal impliceret i risiko for hjertearytmi. Videnskab (2002) 297: 1333-6. doi:10.1126 / videnskab.1073569
CrossRef Fuld Tekst
113. CR, CR. Genetik af hjertearytmier. Hjerte (2005) 91:1352-8.