Genetikken af spaltet læbe og gane
 Intro/abstractvenstre læbe med eller uden ganespalte er en kompleks medfødt anomali, der kan isoleres eller ses sammen med andre misdannelser. Det kan også være en del af fænotypen af et genetisk syndrom. Denne artikel fungerer som en gennemgang af forekomsten af spaltet læbe og gane, risici for gentagelse og risici for andre medfødte anomalier. Genetiske syndromer og teratogene eksponeringer, der vides at være forbundet med orale kløfter, vil blive undersøgt. Derudover genetiske tests, der ofte anmodes om i pædiatrisk klinisk genetik indstilling til evaluering af patienten med spaltet læbe og gane vil blive diskuteret.
Intro/abstractvenstre læbe med eller uden ganespalte er en kompleks medfødt anomali, der kan isoleres eller ses sammen med andre misdannelser. Det kan også være en del af fænotypen af et genetisk syndrom. Denne artikel fungerer som en gennemgang af forekomsten af spaltet læbe og gane, risici for gentagelse og risici for andre medfødte anomalier. Genetiske syndromer og teratogene eksponeringer, der vides at være forbundet med orale kløfter, vil blive undersøgt. Derudover genetiske tests, der ofte anmodes om i pædiatrisk klinisk genetik indstilling til evaluering af patienten med spaltet læbe og gane vil blive diskuteret.
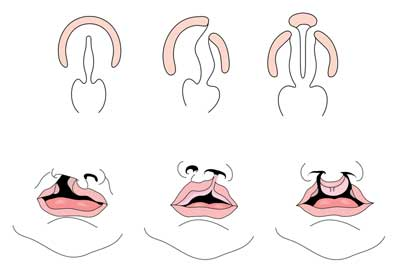
spaltet læbe med eller uden ganespalte (CL/CP) adskiller sig fra en isoleret ganespalte (CP) på embryonale, epidemiologiske og genetiske niveauer. Spaltet læbe skyldes typisk den maksillære prominens og mediale nasale prominens, der ikke smelter sammen mellem den femte og sjette uge med embryonal udvikling. Normal udvikling af ganen skyldes dannelsen af den primære gane og den sekundære gane. Den primære gane dannes i uger seks til syv ved udvikling og fusion af de mediale nasale, laterale nasale og maksillære processer. Den sekundære gane stammer fra de palatale hylder (der udvikler sig fra de parrede maksillære processer i den første grenbue) bliver vandret og smelter sammen og danner de hårde og bløde ganer omkring den niende uge med embryonal udvikling. Hylder smelter også sammen med den primære gane og næseskillevæggen. (1)
orale kløfter er en af de mest almindelige fødselsdefekter, der ses i den nyfødte børnehave, med en samlet prævalens på 1.6 pr. tusind nyfødte over hele verden, med CL/CP set i ca.en pr. tusind fødsler og CP set i 0,6 pr. tusind fødsler. (2) der er en højere frekvens af CL/CP hos personer af asiatisk, afrikansk og indiansk afstamning. CL / CP er også mere almindelig hos mænd. I modsætning hertil er der ingen signifikant forskel i forekomsten af CP blandt forskellige etniske baggrunde, og CP er mere almindelig hos kvinder. (3) risici for gentagelse inden for en familie afhænger af, om kløften er isoleret (uden andre kliniske fund til stede) eller ses som en del af et genetisk syndrom. De fleste tilfælde af orale kløfter er isoleret (ca.80%). Isolerede kløfter menes at have multifaktoriel arv: de skyldes en kombination af flere faktorer, både genetiske og miljømæssige. Risikoen for tilbagefald (tabel 1) øges, når der er mere end en berørt slægtning. Risikoen for gentagelse øges også, jo mere alvorlig defekten er.

Cleft læbe og gane kan ses med andre medfødte anomalier. Sandsynligheden for en genetisk eller teratogen etiologi øger de mere medfødte anomalier, som en patient præsenterer. Tilstedeværelse af andre problemer såsom intellektuel handicap, adfærdsproblemer såsom autisme, dysmorfe træk eller andre medicinske bekymringer vil også gøre en genetisk lidelse eller en teratogen eksponering mere sandsynlig. 13% af personer med spaltet læbe vil have andre medicinske bekymringer eller anomalier. Antallet stiger til 37% med spaltet læbe og gane og til 47% med ganespalte alene.
prænatal eksponering for teratogene stoffer (såsom thalidomid, antikonvulsiva, alkohol, retinsyre og cigaretter) og modersygdom (såsom diabetes, røde hunde og folatmangel) har vist sig at øge risikoen for orale kløfter. Tilstedeværelsen af amniotiske bånd øger også risikoen for kløfter. Periconceptual folinsyretilskud er kendt for at reducere risikoen for orale kløfter.
Pierre Robin-sekvens er en kraniofacial anomali, der er kendetegnet ved mandibulær hypoplasi eller mikrognathia, sekundær U-formet ganespalte og glossoptose, der fører til obstruktiv apnø og fodringsproblemer. Pierre Robin sekvens kan ses som en del af genetiske syndromer (22k11.2 deletion syndrom, Stickler syndrom; beskrevet nedenfor). (5)
der er hundreder af genetiske syndromer forbundet med orale kløfter, herunder cytogenetiske abnormiteter (aneuploidier, mikrodeletioner) og single-gen (Mendelian) lidelser. Bekræftelse af en genetisk diagnose er afgørende for at bestemme prognosen og etablere en risiko for gentagelse.
Aneuploidier som trisomi 13 og 18 har en stærk tilknytning til CL/CP. Trisomi 13 (AKA Patau syndrom) er forbundet med tre kopier af kromosom 13 eller ubalancerede Robertsonian translokationer, der involverer kromosom 13. Babyer født med denne tilstand dør typisk i den nyfødte periode. Kliniske træk inkluderer spaltet læbe og gane, væksthæmning, alvorlige misdannelser i centralnervesystemet (inklusive holoprosencephaly), mikrocephaly, mikropthalmia, iris coloboma, fravær af øjne, misdannede ører, polydactyly, knyttede næver, vippebundsfødder, medfødte hjertefejl og urogenitale defekter. Midtlinjespalter (ellers meget sjældne) kan ses ved trisomi 13 på grund af risikoen for midtlinjedefekter, inklusive holoprosencephaly. Trisomi 18 skyldes typisk tre forskellige kopier af kromosom 18 og er forbundet med dårligt postnatalt resultat. Kliniske træk inkluderer spaltet læbe og gane, intellektuel handicap, manglende trivsel, medfødt hjertesygdom, hypertoni, mikrognathia, kort brystben, misdannede ører med lavt sæt, knyttede hænder, vippebundsfødder og hypoplastiske negle, blandt andre. Trisomi 13 og 18 kan let bekræftes eller udelukkes ved at udføre kromosomanalyse (karyotyping).
Mikrodeletionssyndromer involverer typisk deletion af en del af et kromosom. Disse sletninger kan være for små til at blive detekteret ved standard karyotyping og kan kræve fisk (fluorescens in situ hybridisering) eller mikroarray-teknologi, der skal detekteres. Et velkendt mikrodeletionssyndrom forbundet med ganespalte er 22k11.2 sletningssyndrom (aka Digeorge/Velocardiofacial syndrom). Palatale abnormiteter inklusive velopharyngeal inkompetence, submukosale kløfter, bifid drøbelog ganespalte ses hos 69% af individer med 22k11.2-sletning og kan være en del af Pierre Robin-sekvensen. Andre kliniske fund inkluderer medfødt hjertesygdom, høretab, dysmorfe træk, immunmangel, hypokalcæmi, nyreanomalier, fodringsproblemer, skeletanomalier og psykiatriske lidelser. 10% af tilfældene med 22k11.2-sletningssyndrom antages at være familiært. Sletningen adskiller sig på en autosomal dominerende måde.(6) ulv-Hirschhorn syndrom, som skyldes en sletning i den korte arm af kromosom 4, er også forbundet med orale kløfter (hos 25% Til 50% af de berørte individer). Karakteristiske ansigtstræk (inklusive fremtrædende glabella, der fører til “græsk-krigshjelmudseende”), medfødt hjertesygdom, intellektuel handicap, anfald, manglende trivsel, mikrognathia, præaurikulære tags eller grober og hypodontia kan også ses som en del af tilstanden.(7)
enkeltgenforstyrrelser med orale kløfter inkluderer Stickler syndrom, Treacher Collins syndromog Van der ville syndrom, blandt mange andre. Stickler syndrom er en kollagenforstyrrelse med autosomal dominerende og mindre almindeligt autosomal recessiv arv. Almindelige træk inkluderer ganespalte (set som en del af Pierre Robin-sekvensen eller uden mikrognathia), høretab (sensorineural og ledende), skeletfund (tidlig debut arthritis, spondyloepiphyseal dysplasi), okulære anomalier (høj nærsynethed, glasagtige abnormiteter) og karakteristiske ansigtstræk (med underudvikling af overkæben og næsebroen, midtface retrusion). Genetisk test for Stickler syndrom kan være kompleks, da mutationer i mindst seks gener er blevet beskrevet hos berørte individer. 90% af patienterne med Stickler-syndrom har mutationer i COL2A1-genet og har en autosomal dominerende form for tilstanden.(8) Treacher Collins syndrom er en autosomal dominerende tilstand, der er kendetegnet ved ganespalte med eller uden læbe hos 28% af de berørte individer. Andre abnormiteter inkluderer hypoplasi af de kindben og underkæben, anomalier i det ydre øre, coloboma i det nedre øjenlåg, ledende høretab, fravær af nedre øjenvipper, præaurikulær hårforskydning på kinderne og choanal stenose eller atresi. Diagnosen af Treacher Collins syndrom er baseret på kliniske og radiografiske fund. Mutationer i mindst tre gener er blevet beskrevet, med mutationer i TCOF1 set hos 78% til 93% af patienterne.(9) Van Der Oude syndrom er kendetegnet ved tilstedeværelsen af medfødte, normalt bilaterale, paramedianske fistler i underlæben (grober) eller undertiden små høje med en sinuskanal, der fører fra en slimhinde i læben, og orale kløfter (inklusive CL/CP og CP). Van der er en autosomal dominant tilstand forbundet med mutationer i IRF6-genet (10). Test for enkeltgen-eller multigenbetingelser kræver direkte analyse af genet ved sekventering og/eller deletion/duplikationsanalyse (såsom MLPA).
i betragtning af at genetiske syndromer med spaltet læbe og gane kan være forbundet med aneuploidier, kromosommikrodeletioner/mikroduplikationer eller enkeltgenforstyrrelser, kan genetisk test være en kompliceret proces. En grundig medicinsk historie, en tre-generations stamtavle, en graviditetshistorie og en dysmorfologiundersøgelse af en klinisk genetiker kan afklare det kliniske billede og muliggøre målrettet genetisk test. Nyere teknologier, herunder microarray, giver mulighed for identifikation af små mikrodeletioner og mikroduplikationer, der tidligere er gået glip af standard karyotyping. Desværre fører denne teknik også til identifikation af sletninger og duplikationer af ukendt klinisk betydning, hvilket komplicerer den genetiske rådgivningsproces. Test for enkeltgenforstyrrelser eller Mendelske lidelser kræver klinisk tilgængelighed af genetisk test for det ønskede gen. Det kan også være dyrt, hvis det ikke er dækket af sygesikring. Nye teknologier såsom næste generations sekventering, eksomsekventering eller genomsekventering (kendt samlet som genomiske tests) er nu blevet klinisk tilgængelige. Ved at analysere hundreder til tusinder af gener samtidigt øger disse tests signifikant diagnostisk kraft og udbytte. Sammenlignet med andre teknikker kan disse tests give et svar hurtigere og på en mere omkostningseffektiv måde. På forskningsområdet har eksom-og genomsekventering ført til identifikation af nye gener samt udvidelse af de kliniske træk og spektrum for genetiske mutationer. Som med microarray-teknologi kan genomiske tests detektere syndromer, der ikke er relateret til patientens præsentation og/eller grund til test. I betragtning af de iboende kompleksiteter ved genetisk testning er informeret samtykke nødvendigt.
konklusion
selvom spaltet læbe og gane er en isoleret anomali i de fleste tilfælde, er der en stærk sammenhæng mellem orale kløfter og andre anomalier og genetiske syndromer. En genetisk evaluering foretaget af en klinisk genetiker og en genetisk rådgiver er afgørende for foregribende vejledning og for at bestemme risici for gentagelse. Genetisk testning, som kræver informeret samtykke, kan koordineres og fortolkes under en genetisk evaluering.
Anya Revah, MS, er senior genetisk rådgiver ved Division of Medical Genetics på Maimonides spædbørn og børnehospital i Brooklyn, NY York. Hun er også et aktivt medlem af Maimonides Medical Center og Kings County Hospital kløft læbe og gane tværfagligt Team. Hun har en kandidatgrad i videnskab i genetisk rådgivning fra Boston University i Boston, Massachusetts.
1. Sadler. Langmans medicinske embryologi. Niende Udgave. Sider 390-395.
2. Parker SE, Mai CT, Canfield MA, Rickard R, vil Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. til Det Nationale Netværk til forebyggelse af fødselsdefekter. Opdaterede nationale skøn over fødselsprævalens for udvalgte fødselsdefekter i USA. 2004-2006. Fødselsdefektforskning (del A): klinisk og Molekylær Teratologi 2010;88: 1008-1016.
3. Fraser FC. Genetik af spaltet læbe og ganespalte. Er. J. Hum. Genet. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC, et al. Periconceptuel folatindtagelse efter supplement og fødeindtagelse reducerer risikoen for ikke-syndromisk spaltelæbe med eller uden ganespalte. Forrige Med 2004; 39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Udviklingsmæssige og genetiske perspektiver på Pierre Robin sekvens. Er. J. Med. Genet. 2013; 163C: 295-305.
6. McDonald-McGinn DM, Emanuel BS, Sackai EH. 22k11. 2 Sletningssyndrom. Sept. 23, 1999. . I: Pagon RA, Adam MP, Ardinger HH, et al., editor. Generevisninger . Seattle: universitetet i Seattle; 1993-2014. Tilgængelig fra: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia A, Carey JC, South ST, et al. Ulv-Hirschhorn Syndrom. Apr. 29, 2002. . I: Pagon RA, Adam MP, Ardinger HH, et al., editor. Generevisninger . Seattle: universitetet i Seattle; 1993-2014. Tilgængelig fra: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, Ala-Kokko L. Stickler syndrom. Juni. 9, 2000. . I: Pagon RA, Adam MP, Ardinger HH, et al., editor. Generevisninger . Seattle: universitetet i Seattle; 1993-2014. Tilgængelig fra: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs ad. Treacher Collins Syndrom. Jul. 20, 2004. . I: Pagon RA, Adam MP, Ardinger HH, et al., editor. Generevisninger . København (CPH): Københavns Universitet, København; 1993-2014. Tilgængelig fra: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy S, et al. IRF6-relaterede lidelser. Okt. 30, 2003. . I: Pagon RA, Adam MP, Ardinger HH, et al., editor. Generevisninger . Seattle: universitetet i Seattle; 1993-2014. Tilgængelig fra: http://www.ncbi.nlm.nih.gov/books/NBK1407/.