Mucopolysaccharidoses: tidlige diagnostiske tegn hos spædbørn og børn
Mucopolysaccharidoses (MPS) er en gruppe af klinisk heterogene sygdomme forårsaget af mangler i lysosomale symptomer, der kræves til nedbrydning af glycosaminoglycaner (GAGs).
MPS er karakteriseret ved progressive og systemiske kliniske manifestationer. På trods af deres biokemiske og genetiske heterogenitet deler forskellige typer vigtige kliniske træk i forskellige kombinationer, herunder led-og skeletdysplasi med stivhed (undtagen MPS IV, hvor der er slaphed) og smerter, grove ansigtstræk, hornhindeforklaring, inguinal eller abdominal brok, tilbagevendende øvre luftvejsinfektioner, hjerteklapsygdom, karpaltunnelsyndrom og variabel neurologisk involvering. Disse træk vises normalt i de første måneder af livet for de alvorlige former og i den tidlige barndom for de mere svækkede former, men undervurderes ofte og tages generelt kun i betragtning, når det er tydeligt.
sjældenheden af disse lidelser og variabiliteten i klinisk præsentation fører ofte til en diagnostisk forsinkelse; dette kan variere fra måneder for de alvorlige former til år for de svækkede (se Rigoldi et al. i dette tillæg). På grund af den hurtige progression og det presserende behov for intervention i de alvorlige præsentationer kan selv måneders forsinkelse i diagnosen give katastrofale resultater for patientens fremtidige helbred.
vores mål er i denne gennemgang at understrege de meget tidlige tegn på de alvorlige former, der skal advare børnelægen ved deres første optræden.
hvilke tegn/symptomer kan vi forvente hos børn med svære former for MPS?
symptomer og tegn, der tyder på forskellige former for MPS, er rapporteret i tabel 1 efter alder. I de første 6 måneder af livet kan lyskebrok, unormalt hyppige luftvejsinfektioner, otitis og organomegali ses hos MPS-patienter, især i MPS i (Hurler syndrom), som har den mest tidlige alvorlige begyndelse. Desuden udvikler disse spædbørn fra 6 til 12 måneder meget ofte en gibbus (toraco-lumbal kyphos), hjertemusling, navlestreg, mild hypotoni og vækstforsinkelse . De kan også have det typiske Grove ansigtsudseende (Fig. 1). MPS II, VI og VII kan præsentere med en lignende tidlig fænotype. MPS IV-patienter har en tidlig skeletfænotype med udseende af hoftedysplasi i de første måneder af livet og duebryst (pectus carinatum) i de første 12-18 måneder. MPS II-spædbørn er til stede med en lignende involvering, men med senere begyndelse. I det andet leveår, MPS I Hurler syndrom spædbørn udvikler kognitiv forsinkelse, mens en MPS II-patient kun kan identificeres på grund af overvækst, hyppige luftvejsinfektioner, og flere operationer ; de typiske ansigtstræk vises først senere. Nogle MPS II-patienter udvikler også hyperaktivitet mellem 18 og 24 måneder. I det tredje år af livet er hyperaktivitet og kognitiv forsinkelse let genkendelig i MPS II og MPS III.
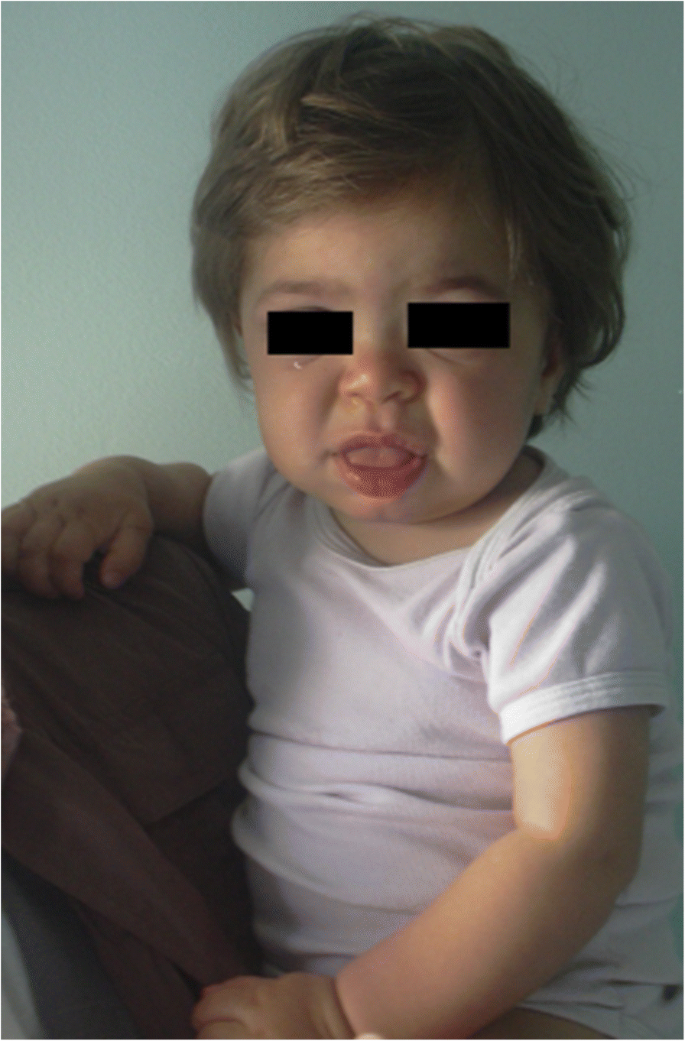
MPS IH i 1-årig patient. Grove facies: flad næsebro, macroglossia, frontal bossing
Sammenfattende er skelet manifestationer typiske og nogle gange for tidlige. Grove facies og kognitiv forsinkelse er ikke tidlige fremtrædende tegn.
hvad er de forskellige præsentationer af de forskellige typer parlamentsmedlemmer?
MPS med somatisk og kognitiv involvering
Mucopolysaccharidosis type i (MPS i; OMIM #252800) (Fig. 1, 2, 3, 4 og 5) er forårsaget af en mangel på lysosomal hydrolase-l-iduronidase, hvilket fører til multisystemakkumulering af to gag ‘ er: dermatansulfat (DS) og heparansulfat (HS) . Baseret på dens sværhedsgrad er MPS i traditionelt opdelt i tre kliniske fænotyper; imidlertid repræsenterer disse former et kontinuum af sygdommens sværhedsgrad. Hurler syndrom, den mest alvorlige form, præsenterer typisk i løbet af det første leveår. Berørte børn udvikler hurtigt signifikant kognitiv svækkelse og somatisk sygdom i flere organsystemer, hvilket fører til død inden for det første årti i fravær af behandling. De svækkede former for MPS i, kendt som Hurler–Scheie syndrom og Scheie syndrom, henholdsvis, er kendetegnet ved senere symptomdebut, længere forventet levetid og mild eller ingen involvering i centralnervesystemet (CNS). Arv er autosomal recessiv, og i mindst 50% af tilfældene er fænotype forudsigelse mulig på basis af genotype, mens der i de resterende 50% opdages nye mutationer . Forekomsten er cirka 1 ud af 100.000 levende fødsler .

MPS IH. en Gibbus i en 9 måneder gammel patient. b rygsøjlen røntgen af rygsøjlen i en 3-årig patient. c T2-vægtet magnetisk resonansbillede af rygsøjlen, der viser signifikant kyphos og vertebral kanalstenose

MPS IH. Røntgenbillede af dysostose multipleks. en fortykkelse af ribbenene hos en 3-årig patient og B hoftedysplasi hos en anden patient i alderen 18 måneder

MPS IH: en klo hånd i en 18 måneder gammel patient; B hånd røntgen.

MPS IH. en fortykkelse af den kortikale knogle i kraniet og unormal “J-formet” konformation af sella turcica. Dilatation af de periventrikulære rum i B FLAIR og c T-2 vægtede magnetiske resonansbilleder af en patient i alderen 17 måneder
Mucopolysaccharidosis type II (MPS II; OMIM #309900) (Fig. 6), også kendt som Hunters syndrom, er resultatet af mangel på iduronat-2-sulfatase (I2S) med deraf følgende GAGAKKUMULERING af DS og HS . Prævalensen er omkring 1 ud af 140.000–156.000 mandlige levende fødsler . Dette er især den eneste MPS, og kvindelige bærere er normalt asymptomatiske; imidlertid, de kan undtagelsesvis påvirkes på grund af abnormiteter i kromosom, homosygositet, eller skæv-inaktivering . Det fænotypiske udtryk spænder også over et bredt spektrum af klinisk sværhedsgrad. I alvorlige tilfælde eksisterer markant progressiv neurologisk involvering og somatisk sygdom, og døden forekommer normalt i det andet årti af livet. Andre patienter med minimal eller ingen neurologisk dysfunktion kan udvise en alvorlig somatisk involvering, mens der i den modsatte ende af spektret er patienter med kun minimale somatiske manifestationer og normal intelligens, der overlever i voksenalderen . Tidlige tegn og symptomer deles af de alvorlige former for MPS I og II.

parlamentsmedlemmer II. En 3-årig patient med kun milde karakteristiske ansigtsegenskaber
sporadiske tilfælde af hydrops fetalis er beskrevet i MPS I , selvom disse børn generelt forekommer normale ved fødslen. De første tegn på MPS i vises normalt i de allerførste måneder af livet, mens de i MPS II normalt forekommer lidt senere . Kiely et al. , i en retrospektiv undersøgelse hos 55 Hurler-patienter, observerede, at åndedrætssymptomer, fodringsvanskeligheder, lyskebrok og otitis media havde vist sig i en medianalder på 6 måneder, og kyphos, hjerteabnormiteter, forstørret hovedomkreds, hypotoni, organomegali, hornhindeforklaring, ledbegrænsning og navlebrok optrådte mellem 6 og 12 måneder. Navlebrok er en af de tidligste præsentere funktioner i MPS I og II, og lyskebrok er rapporteret i cirka 60% af patienterne .
en anden almindelig indledende ledetråd for MPS i og II kan være tilbagevendende infektioner i øvre luftveje med øgede sekretioner. Hyppig otitis, kronisk tilbagevendende rhinitis og vedvarende næseudflod uden åbenbar infektion er ikke specifik, men kan hjælpe med at styrke den diagnostiske mistanke, hvis den er forbundet med mere specifikke tegn. Opbevaring af GAG i oro-svælg fører til forstørrelse af mandler og adenoider og bidrager til komplikationer i de øvre luftveje. Et andet hyppigt fund er støjende vejrtrækning, især om natten, forbundet i de senere stadier med obstruktiv søvnapnø. Som følge af hyppige mellemøreinfektioner og dysostose i mellemørets knogler er høretab hyppigt med både ledende og sensorineurale underskud . Tracheobronchomalacia observeres almindeligvis og kan føre til akut luftvejsobstruktion eller sammenbrud, idet dette er en af hovedårsagerne til døden i de efterfølgende år .
adenoidektomi, tonsillektomi og tympanostomi sammen med brokreparation er de hyppigere og tidlige kirurgiske indgreb for MPS i-og II-patienter, der ofte udføres, før de kender diagnosen (i mere end 50% af tilfældene) .
tilbagevendende diarre er ret almindelig i tidlige stadier af MPS I og kan også være til stede hos MPS II-patienter med neurologisk involvering, mens det er usædvanligt hos de mildt berørte patienter .
Gibbus deformitet (dorso-lumbal kyphosis) (Fig. 2) bliver klinisk synlig i en medianalder på 8.6 måneder til 1 år i MPS I , der repræsenterer en ganske specifik og for tidlig markør for MPS generelt. Hvirvellegemer forekommer fladt og næbbet, hvilket potentielt fører til senere komplikationer såsom rygmarvsindfangning, akut rygmarvsskade og atlanto-occipital ustabilitet; derfor skal der udvises særlig omhu for at forhindre dislokation af det atlanto-aksiale led under generel anæstesi og kirurgi. Efter 1 års alder repræsenterer ledkontrakturer og genu valgum andre hyppige tegn på ledhæmning og progressiv skeletdysplasi, som let kan påvises tidligere end 2 år hos alle spædbørn, der er ramt af MPS. Alle disse skelet manifestationer sammen, typisk for MPS, er navngivet dysostose multipleks. Fortykning af ribbenene kan ses på røntgenbilleder (Fig. 3a), de lange knogler er korte med brede aksler, og knæene er tilbøjelige til valgus og varus deformiteter. Phalangeal dysostose og synovial fortykkelse fører til en karakteristisk klo hånd deformitet (Fig. 4), hvilket er et andet typisk træk, der kan rejse mistanke om sygdommen. Hoftedysplasi er ofte grunden til, at disse børn først kan ses af ortopædiske specialister. Typisk er bækkenet dårligt dannet, lårbenshovederne er små, og coksa valga er almindelig med progressiv og svækkende hoftedeformitet (Fig. 3b).
der er en bemærkelsesværdig forskel mellem MPS i og MPS II; hos MPS i aftager skeletvæksten med 3 år, mens MPS II-patienter viser overvækst i de første 5-6 år , hvilket bremser deres vækst derefter .
grovdannelse af ansigtsegenskaberne (bred næse med blussede næsebor, fremtrædende supraorbitale kamme, store afrundede kinder, tykke læber, forstørret fremspringende tunge) forårsaget af opbevaring af GAGs i det bløde væv i orofacialområdet og ansigtsbenene bliver tydelige inden for de første 2 år, i en medianalder på 1,1 år for Hurler-sygdom og mellem 18 måneder og 4 år i den alvorlige form for Jægersygdom , skønt subtile fænotypiske ændringer kan påvises tidligere (så tidligt som i anden halvdel af det første år) af børnelæger med særlig erfaring (fig. 1). Makrocephaly bliver gradvist mere tydelig, og scaphocefaly er almindelig (Fig. 5), men mikrocefali er også mulig hos Hurler-patienter . Ansigts-og kropshirsutisme er altid tydelig i en alder af 24 måneder . Huden kan være fortykket og uelastisk. Især udvikler nogle MPS II-patienter en markant hudlæsion, der beskrives som elfenben-hvide papler (stenlignende) på øvre ryg og sider af overarmene, patognomonisk af Hunters syndrom .
kardiomyopati og progressiv fortykning og afstivning af ventilbrochurerne hos MPS i-patienter kan føre til mitral og, sjældnere, aorta-regurgitation . En murmur kan påvises i de første måneder af livet og er undertiden rapporteret som det første tegn på sygdommen . Hjerteinddragelse er også til stede hos næsten alle patienter med MPS II, men dette starter omkring 5 år . Ventilsygdom kan føre til højre og venstre ventrikulær hypertrofi og hjertesvigt.
fremspring af maven forårsaget af progressiv hepatosplenomegali er let tydelig efter 6 måneders alder, selvom opbevaring af gag ‘ er i leveren og milten ikke fører til organdysfunktion .
hornhindeforklaring forekommer hos næsten alle personer med MPS i tidligere end 15 måneder , hvilket potentielt forårsager alvorlig synshandicap. Tværtimod er hornhindeforklaring ikke et typisk træk ved Hunters syndrom , men spaltelampeundersøgelse kan afsløre diskrete hornhindelæsioner, der ikke påvirker synet. Retinal dysfunktion kan påvises, mens glaukom ikke er et almindeligt fund.
tidlig psykomotorisk udvikling opdages generelt som normal, men i MPS i er udviklingsforsinkelse normalt åbenlyst i en alder af 18 måneder med en progressiv mental tilbagegang, der fører til en alvorlig intellektuel handicap. Sprogfærdigheder er meget begrænsede hos disse børn. En global forsinkelse af udviklingsmilepæle i MPS II bliver tydelig fra 2 år og fremefter . Imidlertid, det førende neurologiske træk ved svær Jægersygdom er repræsenteret af markante adfærdsforstyrrelser, såsom hyperaktivitet, stædighed, og aggressivitet, der typisk ikke observeres hos dem med den svækkede fænotype .
Mucopolysaccharidosis Type VII (MPS VII; OMIM #253220), også kendt som Sly-syndrom, er en ultra-sjælden MPS, der er kendetegnet ved mangelfuld aktivitet af purpurglucuronidase med lysosomal opbevaring af chondroitinsulfat (CS), DS og HS, hvilket fører til cellulær og organdysfunktion. Den første patient blev beskrevet af Sly et al. i 1973 og i alt 145 patienter er blevet rapporteret i litteraturen indtil nu.
den kliniske præsentation og sygdomsprogression af MPS VII spænder over et bredt sværhedsspektrum, fra tidlige, alvorlige, multisystem manifestationer og død i de allerførste måneder til en mildere fænotype med senere begyndelse, normal eller næsten normal intelligens og længere overlevelse .
Fænotypekarakteristika hos patienter med MPS VII ligner dem hos MPS i og MPS II (kort statur, skeletdysplasi, makrocephaly, tandkødshypertrofi, tilbagevendende øreinfektioner, hepatosplenomegali, brok, hjerteinddragelse, nedsat lungefunktion og kognitiv svækkelse). En uventet høj andel af de beskrevne patienter (41%) har en historie med ikke-immun neonatal hydrops fetalis (NIHF) . På trods af sygdommens prænatale begyndelse hos disse patienter overlevede 13 ud af 23 barndom med et mildt til mellemliggende forløb i deres sene teenageår. Tilstedeværelsen af NIHF forudsiger således ikke i sig selv den mulige sværhedsgrad af det kliniske forløb, hvis patienten overlever spædbarnet. Et andet tidligt tegn er makrokrani, mens hjerteventilinddragelse og gentagne infektioner i øvre og nedre luftveje forekommer i de første 2 leveår. Moderat mental retardation og progressivt høretab med talehæmning er også tydeligt med tiden.
sammenfattende har de alvorlige former for MPS en meget tidlig multisystem debut (inden for 6 måneder af livet); MPS II er ens, men med en senere begyndelse, og MPS VII har en prænatal begyndelse med hyppig NIHF og tidlig alvorlig involvering af den øvre og nedre luftvej.
MPS med for det meste neurologisk og kognitiv involvering
Mucopolysaccharidoser type III (MPS III, også kendt som Sanfilippo syndrom; Fig. 7) har en udbredt neurologisk præsentation. MPS III er en lysosomal opbevaringsforstyrrelse forårsaget af mangel på et af de fire tegn, der er involveret i katabolismen af HS. Fire forskellige undertyper er kendt—MPS III type A (OMIM #252900), type B (omim #252920), type C (omim #252930) og type D (omim #252940)—hver på grund af en anden mangel: type A, sulfamidase/SGSH gen; type B, alfa-n-acetylglucosaminidase/NAGLU gen; Type C, alfa-glucosaminid N-acetyltransferase/hgsnat-gen; type D, N-ACETILGLUCOSAMINA-6-solfato SULFATASI/gns-gen). Alle fire typer (A til D) har autosomal recessiv arv. MPS III er den hyppigste af MPS med en estimeret prævalens mellem 0,3 og 4.1 tilfælde for hver 100.000 nyfødte, afhængigt af undertype og race .

parlamentsmedlemmer IIIA. Den samme patient ved en 2,5 og b 5 år gammel. Bemærk Det forskellige ansigtsudtryk, der viser progression af den neurologiske svækkelse
patienter med Sanfilippo syndrom viser progressiv kognitiv svækkelse i andet og tredje leveår. I modsætning til flertallet af parlamentsmedlemmer er somatiske træk relativt mindre tydelige, og CNS-involveringen er iøjnefaldende. Taleudviklingsforsinkelse er normalt det første tegn, som forældrene bemærker, men de største bekymringer kan være adfærdsproblemer (hyperaktivitet, ængstelig og aggressiv adfærd, søvnforstyrrelser) efterfulgt af progressiv mental tilbagegang . Disse symptomer kan forårsage fejldiagnose som adfærdsmæssige forstyrrelser, opmærksomhedsunderskud hyperaktivitetsforstyrrelse (ADHD) og autismespektrumforstyrrelser . Epilepsi kan også være til stede.
søvnforstyrrelser, hvis forekomst er rapporteret op til 80-90% , er et fælles træk. De består af vanskeligheder med at falde i søvn og hyppig natlig opvågning, med fuldstændig reversering af dag–nat-rytmen hos nogle patienter.
alle disse manifestationer er meget vanskelige at håndtere og har en enorm psykologisk indvirkning på livskvaliteten for hele familien.
selvom fænotypen generelt kan være meget ens i de fire undertyper, synes det kliniske forløb i Sanfilippo B og C generelt at være kendetegnet ved en mindre alvorlig fænotype .
ikke-neurologiske symptomer er normalt mindre udtalte i MPS III end i de andre MPS. Tilbagevendende øre -, næse-og halsinfektioner observeres i en yngre alder. Tilbagevendende otitis, defekter i mellemøret og abnormiteter i det indre øre kan forårsage døvhed. Et overskud af tykke sekretioner og anatomiske ændringer kan producere obstruktion af luftvejene. I de første leveår rapporteres ofte diarre, mens forstoppelse er mere almindelig hos ældre patienter.
fysisk undersøgelse kan vise grove ansigtsegenskaber, selvom disse er mindre udtalt. Patienter har makrocephaly, brede øjenbryn med medial flaring og synophrys, håret er normalt tørt og groft, og hypertrichose er almindeligt. Hos yngre patienter observeres ofte mild hepatomegali og navlebrok og lyskebrok. Sjældne og ofte milde kontrakturer findes for det meste i albuerne. Væksten påvirkes hos ca .halvdelen af patienter over 12 år.
sammenfattende har MPS III eller Sanfilippo sygdom en fremtrædende neurologisk involvering med meget milde somatiske tegn. Småbørn i alderen 2-3 år, der udvikler hyperaktivitet, ADHD og aggressiv adfærd, kan mistænkes for at have MPS III.
MPS med udbredt somatisk involvering
MPS IV og MPS VI er kun til stede med somatisk involvering. Dette er progressive forhold, der skåner intellektuel evne og hovedsageligt påvirker skelettet (MPS IVA) eller alle kroppens organer/systemer (MPS VI). Arv er autosomal recessiv. Den hastighed, hvormed symptomerne forværres, varierer blandt berørte personer.
mucopolysaccharidose IV (MPS IV, også kendt som Morkovo syndrom; Fig. 8) præsenterer som to genetisk forskellige lidelser, hver med en anden mangel: MPS IVA (omim #253000) på grund af mangel på N-acetylgalactosamin-6-sulfatase (GALNS-gen), hvilket resulterer i akkumulering af keratan-sulfat (KS) og CS; og MPS IVB (omim B syndrom ; OMIM #253010) på grund af mangel på beta-galactosidaseaktivitet, der fører til udskillelse af urin KS og oligosaccharid som ved GM1 patienter. MPS IVB er faktisk allelisk for de forskellige former for GM1-gangliosidose, men mangler den psykomotoriske forringelse set i GM1 gangliosidose .

parlamentsmedlemmer IVA. en 4-årig patient med B antero-posterior og C laterale røntgenstråler, der viser “padleformede” ribben og flade og afrundede ryghvirvler med et typisk” forreste næb ” – aspekt
patienter med svær MPS IVA uden påviselig aktivitet afslører deres første symptomer normalt i løbet af det første leveår, selvom de sjældent diagnosticeres før 4-5 år. Oplysninger om den naturlige historie af Morco a patienter er hovedsageligt afledt af to omfattende samlinger af data . Reduceret vækstrate begynder ved cirka 18 måneders alderen, og væksten stopper ved cirka 7 eller 8 år. I modsætning til de andre parlamentsmedlemmer er leddene normalt slappe og meget fleksible (hypermobile), men nogle LED (normalt hofter og skuldre) kan have et begrænset bevægelsesområde. Patienter med sådan skeletinddragelse kan modtage en diagnose af eller gennemgå evaluering for spondyloepiphyseal dysplasi, pseudoachondroplasia, multiple epifysiske dysplasi eller bilateral Legg-Calv Kurrus-Perthes sygdom .
en konstant funktion er odontoid hypoplasi; således kan dislokation af det atlanto-occipitale led forekomme og forårsage kompression og skade på bulbar og rygmarv, hvilket resulterer i lammelse eller endda død, hvis cervikal stabilisering ikke udføres tidligt.
MPS IVB-patienter har en meget lignende klinisk manifestation, men med en mindre fremtrædende kort statur. De har mildt grove ansigtsegenskaber, høretab, hornhindeopaciteter, valvulær hjertesygdom og hyppige øvre luftvejsinfektioner. De kan også præsentere med lyskebrok og mild hepatomegali. Skeletale dysplastiske træk omfatter platyspondyly, odontoid hypoplasi med mulig cervikal subluksation, kypho-skoliose, coksa valga og indsnævrede iliac vinger .
mucopolysaccharidose VI (MPS VI, også kendt som Maroteauks-Lamy syndrom; OMIM #253200; Fig. 9) skyldes patogene mutationer i arylsulfatase B (ARSB) genet placeret på kromosom 5. Som følge heraf forringer reduceret eller fraværende aktivitet af arylsulfatase B (ASB) (N-acetylgalactosamin 4-sulfatase) nedbrydning af DS, der bestemmer dens cellulære akkumulering .

MPS VI. en 2-årig dreng med tydelig grov ansigt og skeletdysostose
patienter uden påviselig aktivitet har den alvorlige form for MPS VI og udviser symptomer i den tidlige barndom. Deres somatiske fænotype ligner Hurler syndrom. I de fleste tilfælde, ved 2 eller 3 år, bliver alvorlig og progressiv skeletskade, der hedder dysostosis multipleks, tydelig. Denne skeletdysplasi inkluderer hoftedysplasi med dysplastisk lårhoved, unormale hvirvellegemer, uregelmæssige kraveben, hypoplastisk distal ulna og radius og dysplastiske og korte metakarpale knogler; karpale og tarsale knogler er hypoplastiske og har en uregelmæssig profil. Væksthastigheden sænkes ofte efter det første leveår med en endelig højde generelt lavere end 120 cm. Som i andre parlamentsmedlemmer er et tidligt tegn grove ansigtsegenskaber. Derudover har patienter en typisk fremspringende mave, hepatomegali, navlestreng og/eller lyskebrok og hjerteinddragelse i den tidlige barndom. Selvom der ikke er nogen primær intellektuel handicap, kan hydrocephalus ses hos nogle patienter med deraf følgende intrakraniel hypertension og papilledem. Pectus carinatum, stive og kontraherede LED, skoliose og kyphos, og cervikal rygmarvskompression forværres med tiden .
sammenfattende viser MPS IV og VI ikke CNS-involvering. MPS IV er en unik MPS i viser fælles slaphed, mens svær MPS VI debut svarer til den observeret i MPS I.