Article
Josivan Gomes Lima1*, Marcel Catão Ferreira dos Santos1, Julliane Tamara Araújo de Melo Campos2
1Departamento de medicina clínica, disciplina de endocrinologia e metabologia. Hospital Universitário Onofre Lopes, Universidade Federal do Rio Grande do Norte (UFRN), Natal, RN, Brasil
2Facultad de Ciencias de la Salud de Trairi, Universidad Federal de Rio Grande do Norte (UFRN), Natal, RN, Brasil
Resumen
La lipodistrofia Generalizada congénita (LCC) es una enfermedad recesiva autosómica rara y grave. Los pacientes tienen defectos en el almacenamiento de grasa corporal y, en consecuencia, depositan grasa en tejidos ectópicos, principalmente en el hígado, y pueden desarrollar cirrosis. La resistencia a la insulina es un hallazgo típico, que causa diabetes que requiere altas dosis diarias de insulina. En el estado de Rio Grande do Norte, Brasil, tenemos una de las cohortes más grandes de pacientes con LCG. En este artículo, revisamos la fisiopatología, el cuadro clínico y el tratamiento de esta enfermedad.
Introducción
La diabetes tipo 2 es un problema de salud mundial, y generalmente es el resultado de un peso excesivo y un aumento de grasa visceral que causan resistencia periférica a la insulina y una incapacidad del páncreas para liberar insulina para compensar esta resistencia. Otros tipos menos comunes de diabetes se producen debido a mutaciones genéticas específicas, como la Lipodistrofia Congénita Generalizada (CGL), también conocida como Lipodistrofia Congénita Berardinelli-Seip (BSCL). La LCC es una enfermedad autosómica recesiva que se clasifica en cuatro tipos, según la mutación genética. Los genes alterados desempeñan funciones esenciales para la formación de adipocitos, la producción de lípidos y el almacenamiento adecuado dentro del adipocito. Las mutaciones disminuyen el tejido adiposo con la consiguiente deposición de grasa en sitios ectópicos, causando hígado graso, metabolismo de carbohidratos alterado, resistencia a la insulina severa con hiperinsulinemia y características acromegaloides, y dislipidemia1-3. El síndrome de CGL tiene alrededor de 500 casos reportados en el mundo. En Brasil, en el Estado de Rio Grande do Norte (RN), hemos diagnosticado, tratado y seguido 54 casos en los últimos 20 años4, 5. En un estudio descriptivo con datos secundarios, se estimó un total de 103 pacientes en RN6. Esto indica una prevalencia mucho mayor que la reportada en la literatura (1: 1 millón) 7.
Formación y almacenamiento de triacilglicerol en gotitas lipídicas
La biosíntesis de triglicéridos y fosfolípidos (Figura 1A) comienza con glicerol-3-fosfato aciltransferasa (GPAT) acilando el glicerol-3-fosfato en la posición 1, formando 1-Acilglicerol-3-fosfato (ácido lisofosfatídico). Es seguido por otro paso de acilación en la posición dos por la enzima AGPAT (1-Acilglicerol-3-fosfato aciltransferasa), que origina 1,2-Diacilglicerol-3-fosfato (ácido fosfatídico). Es un paso intermedio clave en la vía de biosíntesis de triglicéridos y fosfoglicéridos. Hay 11 isoformas de enzimas AGPAT, codificadas por diferentes genes4. AGPAT1 y AGPAT2 son los más estudiados. AGPAT1 está presente en niveles altos en testículos, páncreas y, en menor medida, en tejido adiposo y otros tejidos como corazón, placenta, cerebro, pulmón, mientras que AGPAT2 es abundante en tejido graso. En los siguientes pasos, la enzima citosólica fosfatasa ácida fosfatídica (PAP o lipina) origina 1,2-diacilglicerol, y la 1,2-diacilglicerol aciltransferasa (DGAT) forma triacilglicerol4. El ácido fosfatídico y el diacilglicerol también pueden originar otros fosfolípidos como cardiolipina, fosfatidilinositol y fosfatidilcolina.
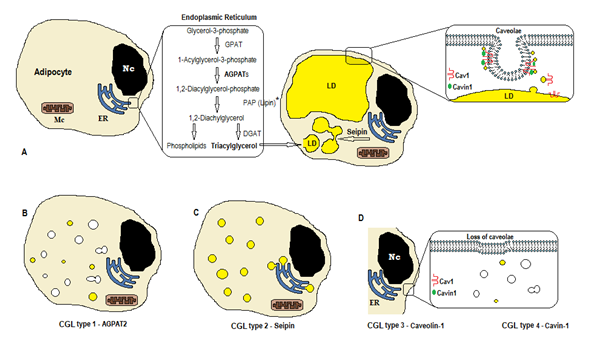
Figura 1. Esquema de síntesis de triglicéridos según tipos de CGL. A) Síntesis y almacenamiento normales de triacilglicerol (TAG) en el adipocito. B) La mutación del AGPAT2 disminuye la producción de TAG (algunos todavía se sintetizan bajo la estimulación de otros AGPAT). C) La mutación del gen seipin disminuye la síntesis de TAG y la formación y fusión de gotitas lipídicas (LD). D) La caveolina-1 y la Cavina-1 son necesarias para la formación y estabilización de las caveolas. La mutación en CAV1 (tipo 3) o CAVIN1 (tipo 4) puede causar pérdida de caveolas en la membrana. Nc, núcleo. Retículo endoplasmático. Mc, mitocondrias. * La lipina es una enzima citosólica anclada por la seipin en la sala de emergencias.
Estas reacciones se producen en el retículo endoplasmático (RE) de los adipocitos, donde una acumulación progresiva de triglicéridos provoca la formación de pequeñas gotitas lipídicas (LD)8. El producto del gen BSCL2 es una proteína transmembrana llamada seipin que causa la fusión de la DL pequeña, originando la DL grande. Seipin reside en el ER y se concentra en la unión con el LD naciente, facilitando el tráfico de lípidos entre el ER y el LD y la incorporación de triglicéridos en el LD9. Seipin también puede actuar como un ancla de ER a la enzima citosólica lipina 1. Además de ser necesario para la fusión, el tamaño y la morfología de las gotas lipídicas, la seipina también es esencial para la adipogénesis (a través de la interacción con la lipina 1) y la lipolisis de triglicéridos celulares10, 11. La deficiencia de seipin dificulta la diferenciación de preadipocitos a adipocitos y afecta a la maduración final9, como lo demuestran los estudios en células madre mesenquimales con BSCL2 eliminado12. Los tejidos no adiposos también expresan seipin, y otras funciones deben determinarse.
En los adipocitos, las caveolas, que son invaginaciones de membrana especializadas de 50-100 nm, representan el 20% del área de la membrana plasmática, lo que hace que los adipocitos sean las células con mayor densidad de caveolas13. La formación de gotitas lipídicas necesita una proteína de membrana (Caveolina, el componente principal de las membranas de caveolas) y una proteína citoplasmática (Cavin – 1)14. Los genes CAV1, CAV2 y CAV3 codifican tres formas de caveolina con estructuras similares (Caveolina-1, Caveolina-2 y Caveolina-3, respectivamente). La caveolina-1 y la caveolina-2 están presentes en adipocitos, fibroblastos y células endoteliales, y la caveolina-3 está presente solo en los músculos esqueléticos y cardiacos13, 15. La caveolina-1 es la más importante y la más estudiada. Se expresa en dos isoformas diferentes (1a y 1b). La caveolina-1 se traslada de la membrana plasmática a una gota lipídica, siendo necesaria para el tráfico y el metabolismo16 de lípidos. Las gotitas lipídicas almacenan triglicéridos después de la alimentación y estas moléculas se hidrolizan en ácidos grasos y se liberan durante el ayuno; este mecanismo puede ser regulado por la Caveolina-116. La deficiencia de caveolina-1 también aumenta la susceptibilidad a la muerte celular por autofagia17.
El gen CAVIN1 codifica una proteína citoplasmática llamada proteína asociada a caveolas 1 (Cavin-1)14, 16, que es obligatoria para la formación y estabilización de caveolas. Cavin-1 se expresa en adipocitos, células musculares y otras células, y también es esencial en la transmisión de signos originados en caveolas 14,18. El knockout del gen CAV1 causa una falta de caveolas en células no musculares, mientras que el knockout de CAVIN1 causa la ausencia de caveolas en todos los tejidos, incluido el musculo14. La falta de caveolas puede afectar la regulación de la lipólisis, el flujo de ácidos grasos, la síntesis de triglicéridos y las señales de otras vías.
Tipos de LCC
En base a alteraciones genéticas detectables, se describen cuatro tipos. Los tipos 1 y 2 son responsables de más del 95% de los casos, y el tipo 2 tiene un fenotipo más gravemente afectado. Solo se ha notificado un caso de tipo 3 y alrededor de 30 casos de tipo 44.
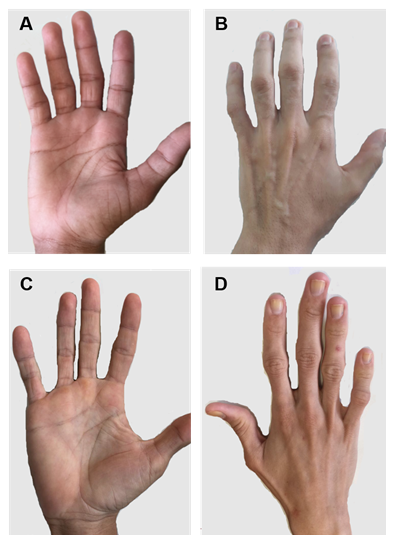
Figura 2. Manos de pacientes con LCG tipos 1 y 2. (A) y (B) Vistas anteriores y posteriores de las manos de pacientes tipo 1. Manos aparentemente normales, ya que todavía hay tejido graso mecánico. (C) y (D) Vistas anteriores y posteriores de las manos de pacientes tipo 2. La gravedad de la enfermedad es mayor, y la falta de grasa es evidente y fácilmente perceptible.
CGL Tipo 1. En 1999, Garg et al. describieron la mutación de los pacientes en el cromosoma 9q34, y tres años después Agarwal et al. mostró AGPAT2 como la enzima afectada por esta mutación2, 19. Debido a la mutación de este AGPAT2, la producción mínima o nula de triacilglicerol ocurre por el estímulo de otras isoformas. El fenotipo de los ratones knockout AGPAT2 es similar al de los humanos con tipo CGL, confirmando el papel de esta enzima en la fisiopatología20, 21.
CGL Tipo 2. Magre et al. fueron los primeros en identificar la mutación en el gen seipin (cromosoma 11q13) 3. Las mutaciones (en su mayoría sin sentido) del gen seipin (BSCL2) producen una proteína truncada y pueden afectar el metabolismo de los lípidos por diferentes mecanismos: a) disminución de la estabilidad de seipin; b) reducción de la capacidad de unirse a la lipina 1; y c) falla en oligomerizarse y localizarse exclusivamente a la membrana de ER11. Algunas células todavía son capaces de generar triacilglicerol y pequeñas gotas de lípidos, pero las grandes gotas de lípidos están ausentes debido a la pérdida de la capacidad de fusión de estas pequeñas gotas de lípidos. También hay una falla en la expresión de factores adipogénicos, como el receptor gamma activado por proliferador de peroxisomas (PPARG), así como la adiponectina y la proteína de unión a ácidos grasos de adipocitos (FABP4)11, 16. La deficiencia de seipin afecta la adipogénesis, aumenta la lipólisis y previene la acumulación de triglicéridos en los adipocitos.
CGL tipo 3. Este tipo fue descrito recientemente en un paciente que a pesar de tener fenotipo CGL no tenía mutaciones en los genes AGPAT2 o BSCL222. Los ratones con una mutación en Cav1 son resistentes a la obesidad inducida por la dieta y tienen resistencia a la insulina, hipertrigliceridemia, disminución de adiponectina, reducción de la masa grasa y pequeños adipocitos16. Después de elegir genes candidatos basados en estudios en ratones, Kim et al. se confirmó la presencia de una mutación sin sentido en el gen caveolina-1 (CAV1), en el cromosoma 7q3122.
CGL tipo 4. En este tipo raro, el gen afectado es el CAVIN1, que codifica la proteína Cavin-1. En humanos, se ha descrito en pacientes con lipodistrofia congénita generalizada y distrofía muscular15, 23.
Recientemente, también se han descrito mutaciones en los genes PCYT1A y PPARG que causan lipodistrofia24, 25.
Características clínicas
Los pacientes con LCG suelen presentar facies acromegaloides, acantosis nigricans, febomegalia, hepatomegalia e hipertrofía muscular5, 26, 27. Varios autores citan la hernia umbilical como un hallazgo clínico del síndrome26. Evaluamos la frecuencia de la misma en nuestra serie de pacientes, y ninguno de ellos presentó este cambio28. De hecho, la ausencia de tejido adiposo periumbilical provoca la protuberancia de la cicatriz umbilical, que puede ser diagnosticada erróneamente como hernia28, 29.
Una vez que los adipocitos no pueden almacenar adecuadamente la grasa, se acumula en otros tejidos, como el hígado y los músculos, causando una severa resistencia a la insulina. La densitometría ósea (DXA) puede mostrar densidad mineral ósea normal o alta 30 y reducción de la grasa corporal total (generalmente inferior al 6%)27. Como consecuencia del bajo nivel de grasa corporal, la adiponectina y la leptina séricas son bajas27. Como la leptina es esencial para controlar el hambre, estos pacientes suelen tener hiperfagia, que es evidente desde la infancia. La adiponectina desempeña un papel importante como sensibilizador de la insulina, y su falta empeora la resistencia a la insulina. A pesar de esto, inicialmente, la glucosa y la hemoglobina glucosilada son normales a expensas de niveles de insulina muy altos. La diabetes suele comenzar en la pubertad; en nuestra serie, la edad media de aparición fue de 15,8±7,1 años27. Inicialmente, se controlan con medicamentos orales, necesitando altas dosis de insulina en pocos años27. La hipertensión arterial ocurre en un tercio de los pacientes27.
Hay algunas características clínicas específicas de cada tipo de LCC. Los pacientes con tipo 1 todavía presentan grasa adiposa mecánica, especialmente en palmas, plantas, regiones orbitales y periarticulares31. Por el contrario, los pacientes de tipo 2 muestran ausencia de tejidos grasos metabólicos y mecánicos. Seipin se expresa altamente en el cerebro y el cerebelo y también participa en la regulación de las funciones neuronales. Más de la mitad de los pacientes de tipo 2 tienen algún deterioro cognoscitivo1, 8. Los tipos 3 y 4 tienen preservación de grasa mecánica y de médula ósea, y el tipo 4 tiene debilidad muscular asociada con creatina-cinasa sérica alta e inestabilidad espinal 15.
También hay características clínicas específicas de género. Los ovarios poliquísticos y la amenorrea son comunes32. Los ciclos menstruales suelen volver a la normalidad con el uso de metreleptina, probablemente debido a la mejora de la sensibilidad a la insulina y al restablecimiento de la pulsatilidad32 de la HL. Los hombres de tipo 2 pueden tener teratozoospermia debido a la falta de seipin en las células germinales33.
La hipertrigliceridemia se produce desde los primeros años de vida y puede causar pancreatitis aguda. El HDL suele ser inferior a 30 mg/dL. Las elevaciones de enzimas hepáticas también son un hallazgo temprano y provienen de la deposición de grasa en el hígado. Las reducciones progresivas de plaquetas en suero sugieren un empeoramiento de la enfermedad hepática y una cirrosis probable34.
Como Cavin-1 está presente en las células musculares, los pacientes con tipo 4 tienen debilidad muscular leve y creatina-cinasa elevada15.
La esperanza de vida, principalmente en el tipo 2, disminuye sustancialmente, y la muerte ocurre con frecuencia antes de los 30 años (experiencia personal basada en 20 pacientes que murieron en los últimos 19 años). Las causas de muerte están relacionadas con diabetes (insuficiencia renal, muerte súbita), hígado (cirrosis, sangrado digestivo) o infecciones.
Diagnóstico y tratamiento
El diagnóstico de CGL se basa en datos clínicos: características acromegaloides, acantosis nigricans, reducción de la grasa corporal total, hipertrofia muscular y protrusión de la cicatriz umbilical. Además, los datos de laboratorio pueden mostrar diabetes con resistencia a la insulina severa e hipertrigliceridemia. Las pruebas por imágenes pueden ayudar a identificar depósitos ectópicos de grasa, principalmente en el hígado y el páncreas (esteatosis hepática con hepatomegalia y esteatosis pancreática). La DXA puede confirmar el bajo nivel de grasa corporal y la alta densidad ósea30.
El tratamiento de CGL consiste en un control estricto de la dieta con la disminución de la ingesta de grasas, principalmente, triglicéridos y alimentos con un alto índice glucémico para prevenir y controlar las comorbidas29. Sin embargo, la dieta ideal es un objetivo difícil de lograr debido al aumento del apetito y la restricción severa preconizada. También se debe fomentar la actividad física para mejorar el control de las comorbilidades, excepto en aquellos pacientes con contraindicaciones como miocardiopatía29 grave.
En cuanto al tratamiento farmacológico, estos pacientes pueden ser tratados con los medicamentos habituales para la diabetes, la hipertensión y la dislipidemia. La primera opción para el tratamiento de la diabetes y la resistencia a la insulina es la metformina, pero generalmente no es suficiente. A diferencia del tratamiento de la lipodistrofia parcial, las tiazolidindionas deben utilizarse con precavida29. Se utilizan otros antidiabéticos orales, pero no se estudiaron específicamente en pacientes con LCC. Hay datos en animales que sugieren que el uso de inhibidores de SGLT2 (dapagliflozina) podría tener beneficios para prevenir la miocardiopatía35; se necesitan estudios para confirmar esto en seres humanos. A medida que la enfermedad progresa y se produce una resistencia grave a la insulina, se necesitan dosis altas diarias de insulina. La falta de tejido adiposo subcutáneo es un problema en la administración de altas dosis de insulina. Puede ser necesaria insulina más concentrada (U-300 o U500) 36. Estos pacientes presentan dislipidemia severa, debido principalmente al aumento de triglicéridos y HDL bajo, y por lo tanto, el uso de fibrato a veces es necesario para prevenir la pancreatitis aguda. Además, debido al alto riesgo cardiovascular de estos pacientes, se debe considerar la intervención con estatinas, y las dianas de LDL o no HDL deben ser estrictas 29.
Las inyecciones diarias de metreleptin causan una disminución significativa del apetito y traen beneficios al reducir la glucemia, la triglicérida y las enzimas hepáticas. Es notable, especialmente en niños, la reducción de la circunferencia abdominal, probablemente debido a una reducción de la hepatomegalia.
Conclusión
El LCC es una enfermedad rara y grave que puede ocurrir con diabetes (que generalmente requiere dosis altas de insulina) y muerte prematura. El fenotipo del paciente es bastante característico, requiriendo, sin embargo, el conocimiento del síndrome por parte de los profesionales de la salud para realizar un diagnóstico precoz. La metreleptina parece ser el único medicamento en este momento que puede modificar la historia natural de la enfermedad.
Conflicto de intereses: ninguno.
- Nolis T. Explorando la fisiopatología detrás de las lipodistrofias genéticas y adquiridas más comunes. Journal of human genetics. 2014 Jan; 59 (1): 16-23.
- Agarwal AK, Arioglu E, De Almeida S, et al. AGPAT2 está mutado en lipodistrofia generalizada congénita ligada al cromosoma 9q34. Nat Genet. 2002 May; 31(1): 21-3.
- Magre J, Delepine M, Khallouf E, et al. Identificación del gen alterado en la lipodistrofia congénita Berardinelli-Seip en el cromosoma 11q13. Genética de la naturaleza. 2001 Aug; 28 (4): 365-70.
- Patni N, Garg A. Lipodistrofias congénitas generalizadas new nuevos conocimientos sobre la disfunción metabólica. Nature revisa Endocrinología. 2015 Sep; 11( 9): 522-34.
- Garg A. Lipodistrofias adquiridas y heredadas. The New England journal of medicine (en inglés). 2004 Mar 18; 350 (12): 1220-34.
- de Azevedo Medeiros LB, Candido Dantas VK, Craveiro Sarmento AS, et al. Alta prevalencia de Lipodistrofia Congénita Berardinelli-Seip en el Estado de Rio Grande do Norte, Nordeste de Brasil. Diabetol Metab Syndr. 2017; 9: 80.
- Chiquette E, EA Oral, Garg A, et al. Estimación de la prevalencia de lipodistrofia generalizada y parcial: hallazgos y desafíos. Diabetes, Síndrome Metabólico y Obesidad: Dianas y Terapia. 2017: 375-83.
- Wee K, Yang W, Sugii S, et al. Hacia una comprensión mecanicista de la lipodistrofia y las funciones del seipin. Bioscience reports (en inglés). 2014; 34(5).
- Dollet L, Magre J, Cariou B, et al. Función de seipin: nuevos conocimientos de los modelos de ratón knockout de Bscl2 / seipin. Biochimie. 2014 Jan; 96: 166-72.
- Sim MF, Dennis RJ, Aubry EM, et al. La proteína de lipodistrofia humana seipin es un adaptador de membrana ER para la fosfatasa adipogénica PA lipina 1. Metabolismo molecular. 2012; 2(1): 38-46.
- Sim MF, Talukder MM, Dennis RJ, et al. El análisis de mutaciones naturales en la proteína de lipodistrofia humana seipin revela múltiples mecanismos patogénicos potenciales. Diabetologia. 2013 Nov; 56(11): 2498-506.
- Payne VA, Grimsey N, Tuthill A, et al. El gen de lipodistrofia humana BSCL2/seipin puede ser esencial para la diferenciación normal de adipocitos. Diabetes. 2008 Aug; 57 (8): 2055-60.
- Cohen AW, Hnasko R, Schubert W, et al. Papel de las caveolas y caveolinas en la salud y la enfermedad. Revisiones fisiológicas. 2004 Oct; 84 (4): 1341-79.
- Pilch PF, Liu L. Cuevas de grasa: caveolas, tráfico de lípidos y metabolismo de lípidos en adipocitos. Tendencias en endocrinología y metabolismo: TEM. 2011 Aug; 22 (8): 318-24.
- Hayashi YK, Matsuda C, Ogawa M, et al. Las mutaciones de PTRF en humanos causan una deficiencia secundaria de caveolinas que resulta en distrofia muscular con lipodistrofia generalizada. J Clin Invest. 2009 Sep; 119( 9): 2623-33.
- Parton RG, del Pozo MA. Caveolas como sensores de membrana plasmática, protectores y organizadores. Nature revisa la biología celular molecular. 2013 Feb; 14 (2): 98-112.
- Le Lay S, Briand N, Blouin CM, et al. El modelo de ratón con deficiencia de caveolina-1 lipoatrófica revela autofagia en adipocitos maduros. Autofagia. Agosto de 2010; 6 (6): 754-63.
- Liu L, Brown D, McKee M, et al. La deleción de Cavin / PTRF causa pérdida global de caveolas, dislipidemia e intolerancia a la glucosa. Metabolismo celular. 2008 Oct; 8 (4): 310-7.
- Garg A, Wilson R, Barnes R, et al. Un gen para la lipodistrofia generalizada congénita se asigna al cromosoma humano 9q34. The Journal of clinical endocrinology and metabolism (en inglés). 1999 Sep; 84 (9): 3390-4.
- Vogel P, Léase R, Hansen G, et al. Patología de lipodistrofia congénita generalizada en ratones Agpat2 -/ -. Patología veterinaria. 2011 May; 48 (3): 642-54.
- Cortes VA, Curtis DE, Sukumaran S, et al. Mecanismos moleculares de esteatosis hepática y resistencia a la insulina en el modelo de ratón deficiente en AGPAT2 de lipodistrofia generalizada congénita. Metabolismo celular. 2009 Feb; 9 (2): 165-76.
- Kim CA, Delepine M, Boutet E, et al. Asociación de una mutación homocigótica sin sentido de caveolina-1 con lipodistrofia congénita Berardinelli-Seip. J Clin Endocrinol Metab. 2008 Apr; 93 (4): 1129-34.
- Rajab A, Straub V, McCann LJ, et al. Arritmia cardíaca fatal y síndrome de QT largo en una nueva forma de lipodistrofia generalizada congénita con ondulación muscular (CGL4) debido a mutaciones en PTRF-CAVIN. PLoS genetics. 12 de marzo de 2010; 6 (3): e1000874.
- Payne F, Lim K, Girousse A, et al. Mutaciones que alteran la vía de fosfatidilcolina Kennedy en humanos con lipodistrofia congénita y enfermedad del hígado graso. Proc Natl Acad Sci U S A. 2014 Jun 17; 111 (24): 8901-6.
- Dyment DA, Gibson WT, Huang L, et al. Las mutaciones bialélicas en PPARG causan una lipodistrofia congénita generalizada similar al síndrome de Berardinelli-Seip. Eur J Med Genet. 2014 Sep; 57( 9): 524-6.
- Garg A. Revisión clínica#: lipodistrofias: trastornos genéticos y de la grasa corporal adquirida. The Journal of clinical endocrinology and metabolism (en inglés). 2011 Nov; 96(11): 3313-25.
- Lima JG, Nobrega LH, de Lima NN, et al. Datos clínicos y de laboratorio de una gran serie de pacientes con lipodistrofia generalizada congénita. Diabetol Metab Syndr. 2016; 8: 23.
- Lima GJ, Lima NN, Oliveira CF, et al. Hernia Umbilical en Pacientes con Síndrome de Berardinelliseip: Es Realmente una Hernia. J Clin Mol Endocrinol. 2015; 1(1): 3.
- Brown RJ, Araujo-Vilar D, Cheung PT, et al. The Diagnosis and Management of Lipodistrophy Syndromes: A Multi-Society Practice Guideline (en inglés). J Clin Endocrinol Metab. Diciembre de 2016; 101( 12): 4500-11.
- Lima JG, Nobrega LH, Lima NN, et al. La Densidad Ósea en Pacientes Con Lipodistrofia Congénita de Berardinelli-Seip Es Mayor en las Localizaciones Trabeculares y en los Pacientes de Tipo 2. J Clin Densitom. 25 de noviembre de 2016.
- Simha V, Garg A. Heterogeneidad fenotípica en la distribución de la grasa corporal en pacientes con lipodistrofia generalizada congénita causada por mutaciones en los genes AGPAT2 o seipin. J Clin Endocrinol Metab. 2003 Nov; 88(11): 5433-7.
- Musso C, Cochran E, Javor E, et al. El efecto a largo plazo del tratamiento con leptina humana metionil recombinante sobre el hiperandrogenismo y la función menstrual en mujeres y en la función hipofisaria en pacientes lipodistróficos hipoleptinémicos masculinos y femeninos. Metabolismo. 2005 Feb; 54 (2): 255-63.
- Jiang M, Gao M, Wu C, et al. La falta de seipin testicular causa el síndrome de teratozoospermia en los hombres. Proc Natl Acad Sci U S A. 2014 May 13; 111 (19): 7054-9.
- Mitchell O, Feldman DM, Diakow M, et al. La fisiopatología de la trombocitopenia en la enfermedad hepática crónica. Hepat Med. 2016; 8: 39-50.
- Joubert M, Jagu B, Montaigne D, et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes. 2017 Apr; 66(4): 1030-40.
- Lima JG, Lima NN, Lima RLM, et al. Glargine U300 Insulin as a Better Option than Degludec U100 to Treat a Congenital Generalized Lipodystrophy Patient. Clin Diabetes Res. 2017; 1(1).