artikkeli
Josivan Gomes Lima1*, Marcel Catão Ferreira dos Santos1, Julliane Tamara Araújo de Melo Campos2
1departamento de medicina clínica, disciplina de endocrinologia e metabologia. Hospital Universitário Onofre Lopes, Universidade Federal do Rio Grande do Norte (UFRN), Natal, RN, Brazil
2terveystieteiden tiedekunta Trairi, Federal University of Rio Grande do North (UFRN), Natal, RN, Brazil
Abstrakti
synnynnäinen yleistynyt lipodystrofia (CGL) on harvinainen ja vaikea autosomaalinen resessiivinen sairaus. Potilaat ovat puutteellisia kehon rasvan varastoinnissa ja näin ollen he tallettavat rasvaa kohdunulkoisiin kudoksiin, pääasiassa maksaan, ja voivat kehittyä kirroosiksi. Insuliiniresistenssi on tyypillinen löydös, joka aiheuttaa diabetesta, joka vaatii suuria vuorokausiannoksia insuliinia. Rio Grande do Norten osavaltiossa Brasiliassa on yksi suurimmista CGL-potilaiden kohorteista. Tässä artikkelissa tarkastelemme patofysiologiaa, kliinistä kuvaa ja tämän taudin hoitoa.
Johdanto
tyypin 2 diabetes on maailmanlaajuinen terveysongelma, ja se johtuu yleensä ylipainosta ja viskeraalisen rasvan lisääntymisestä, mikä aiheuttaa perifeeristä insuliiniresistenssiä ja haiman kyvyttömyydestä vapauttaa insuliinia tämän resistenssin kompensoimiseksi. Muita harvinaisempia diabetes johtuu erityisiä geneettisiä mutaatioita, kuten synnynnäinen yleistynyt lipodystrofia (CGL), joka tunnetaan myös nimellä Berardinelli-Seip synnynnäinen lipodystrofia (Bscl). CGL on autosomaalinen resessiivinen sairaus, joka luokitellaan geenimutaation perusteella neljään tyyppiin. Muutetuilla geeneillä on tärkeitä tehtäviä rasvasolujen muodostukselle, lipidituotannolle ja asianmukaiselle varastoinnille rasvasolujen sisällä. Mutaatiot vähentävät rasvakudosta ja sitä kautta rasvakudosta kohdunulkoisiin kohtiin aiheuttaen rasvamaksaa, muuttunutta hiilihydraattiaineenvaihduntaa, vaikeaa insuliiniresistenssiä, johon liittyy hyperinsulinemiaa ja akromegaloidiominaisuuksia, sekä dyslipidemia1-3. CGL-oireyhtymästä on raportoitu maailmalla noin 500 tapausta. Brasiliassa, Rio Grande do Norten osavaltiossa (RN), olemme diagnosoineet, hoitaneet ja seuranneet 54 tapausta viimeisten 20 vuoden aikana 4, 5. Kuvailevassa tutkimuksessa, jossa käytettiin toissijaisia tietoja, arvioimme yhteensä 103 potilasta RN6: ssa. Tämä osoittaa, että esiintyvyys on paljon suurempi kuin kirjallisuudessa on ilmoitettu (1: 1 miljoonaa) 7.
Triasyyliglyserolin muodostuminen ja varastointi lipidipisaroissa
triglyseridien ja fosfolipidien biosynteesi (Kuva 1a) alkaa, kun glyseroli-3-fosfaattiasyylitransferaasi (GPAT) asyloi glyseroli-3-fosfaatin asemassa 1 muodostaen 1-Asyyliglyseroli-3-fosfaattia (lysofosfatidihappoa). Sitä seuraa toinen asylointivaihe kohdassa kaksi 1,2-Diasyyliglyseroli-3-fosfaattiasyylitransferaasientsyymi AGPAT (1-Asyyliglyseroli-3-fosfaatti), joka on peräisin 1,2-Diasyyliglyseroli-3-fosfaatista (fosfatidihappo). Se on tärkeä välivaihe sekä triglyseridien että fosfoglyseridien biosynteesireitissä. Agpat-entsyymejä on 11 isoformia, joita koodaa eri genes4. AGPAT1 ja AGPAT2 ovat laajimmin tutkitut. AGPAT1: tä esiintyy runsaasti kiveksissä, haimassa ja vähemmässä määrin rasvakudoksessa ja muissa kudoksissa, kuten sydämessä, istukassa, aivoissa ja keuhkoissa, kun taas agpat2: ta on runsaasti rasvakudoksessa. Seuraavissa vaiheissa sytosolientsyymi fosfatidihappofosfataasi (PAP tai lipiini) valmistaa 1,2-diasyyliglyserolia ja 1,2-diasyyliglyseroliasyylitransferaasi (DGAT) muodostaa triasyyliglyseroli4. Fosfatidihappo ja diasyyliglyseroli voivat olla peräisin myös muista fosfolipideistä, kuten kardiolipiinista, fosfatidyylinositolista ja fosfatidyylikoliinista.
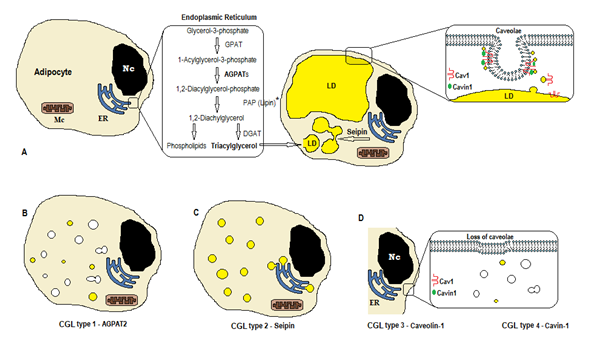
Kuva 1. Järjestelmän triglyseridien synteesin mukaan CGL tyyppejä. (A) normaali triasyyliglyserolin (TAG) synteesi ja varastointi rasvasoluissa. (B) AGPAT2: n mutaatio vähentää tagin tuotantoa (osa syntetisoidaan edelleen muiden Agpat: iden stimulaatiolla). (C) mutaatio seipin geenin vähentää TAG synteesi ja lipidipisaran (LD) muodostumista ja fuusio. (D) Kaveolin-1: tä ja Cavin-1: tä tarvitaan luolien muodostamiseen ja stabilointiin. Mutaatio CAV1: ssä (tyyppi 3) tai CAVIN1: ssä (tyyppi 4) voi aiheuttaa caveolae: n katoamista kalvossa. NC, nucleus. Endoplasma reticulum. Mc, mitokondriot. * Lipin on sytosolinen entsyymi, joka ankkuroituu SEIPINIIN ER: ssä.
nämä reaktiot tapahtuvat adiposyyttien endoplasmaisessa retikulumissa (ER), jossa triglyseridien asteittainen kertyminen aiheuttaa pienten lipidipisaroiden (LD)muodostumisen 8. Geenin bscl2 tuote on transmembraaniproteiini nimeltä seipiini, joka aiheuttaa pienen LD: n fuusion, joka on peräisin suuresta LD: stä. Seipiini sijaitsee ER: ssä ja konsentroituu orastavan LD: n liittymäkohdassa, mikä helpottaa LIPIDILIIKENNETTÄ ER: n ja LD: n välillä ja triglyseridien liittymistä LD9: ään. Seipiini voi toimia myös SYTOSOLISEN entsyymin lipiini 1: n ankkurina. Sen lisäksi, että seipiini on välttämätön lipidipisaroiden fuusiolle, koolle ja morfologialle, seipiini on myös välttämätöntä adipogeneesin (vuorovaikutuksen kautta lipiini 1: een) ja solujen triglyseridien lipolysis10, 11. Seipiinin puute haittaa esiadiposyyttien erilaistumista adiposyyteiksi ja vaikuttaa lopulliseen kypsymiseen9, kuten mesenkymaalisilla kantasoluilla tehdyt tutkimukset bscl2: lla ovat osoittaneet 12. Myös muut kuin rasvakudokset ilmaisevat seipiiniä, ja muut toiminnot on määritettävä.
adiposyyteissä 50-100 nm: n kalvohyökkäyksiin erikoistuneiden luolien osuus plasman kalvopinta-alasta on 20%, jolloin adiposyyteissä on eniten kaveoleja 13. Lipidipisaroiden muodostuminen tarvitsee kalvoproteiinia (Kaveoliini – kaveolae-kalvojen pääkomponentti) ja sytoplasmaproteiinia (Cavin-1)14. Geenit CAV1, CAV2 ja CAV3 koodaavat kolmea kaveoliinin muotoa, joilla on samanlaiset rakenteet (Caveolin-1, Caveolin-2 ja Caveolin-3). Kaveolin-1 ja Kaveolin-2 ovat läsnä adiposyyteissä, fibroblastissa ja endoteelisoluissa, ja Kaveolin-3 on läsnä vain luuston ja sydämen lihaksissa 13, 15. Caveolin – 1 on tärkein ja tutkituin. Se ilmaistaan kahtena eri isoformina (1a ja 1b). Kaveolin-1 translokoituu plasmakalvosta lipidipisaraksi, mikä on välttämätöntä lipidien kulkeutumiselle ja metabolismille16. Lipidipisarat varastoivat triglyseridejä ruokinnan jälkeen ja nämä molekyylit hydrolysoituvat rasvahapoksi ja vapautuvat paaston aikana; tätä mekanismia voidaan säädellä Caveolin-116: lla. Caveolin-1-puutos lisää myös alttiutta solukuolemalle autofagi17.
geeni CAVIN1 koodaa sytoplasmaproteiinia nimeltä caveolae associated protein 1 (Cavin-1)14, 16, joka on pakollinen caveolae: n muodostumiselle ja stabiloinnille. Cavin-1 ilmaistaan adiposyyteissä, lihassoluissa ja muissa soluissa, ja se on myös välttämätön caveolae-alkuisten signals14, 18-solujen välittämisessä. Cav1-geenin tyrmäys aiheuttaa caveolien puutteen muissa kuin lihassoluissa, kun taas cavin1: n tyrmäys aiheuttaa caveolien puuttumisen kaikista kudoksista, myös lihaksista14. Caveolae puuttuminen voi vaikuttaa lipolyysin, rasvahappovirran, triglyseridien synteesin ja muiden reittien signaalien säätelyyn.
CGL: n tyypit
havaittavien geneettisten muutosten perusteella on kuvattu neljää tyyppiä. Tyypit 1 ja 2 ovat vastuussa yli 95% tapauksista, ja tyypin 2 on vakavammin vaikuttanut fenotyyppi. Vain yksi tyypin 3 tapaus ja noin 30 tyypin 4 tapausta on ilmoitettu 4.
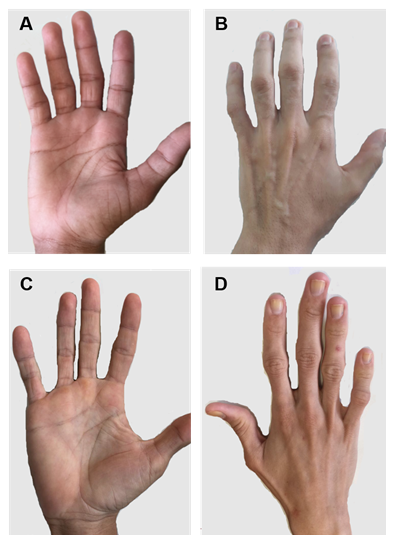
kuva 2. CGL-tyyppien 1 ja 2 potilaiden kädet. A) ja B) tyypin 1 potilaiden käsien Anterior-ja posteriorinäkymät. Ilmeisesti normaalit kädet, koska siellä on vielä mekaanista rasvakudosta. C) ja D) tyypin 2 potilaiden käsien Anterior-ja posteriorinäkymät. Taudin vakavuus on suurempi, ja rasvan puute on ilmeinen ja helposti havaittavissa.
CGL-tyyppi 1. Vuonna 1999 Garg et al. kuvattu potilaiden mutaatio kromosomissa 9q34, ja kolme vuotta myöhemmin Agarwal et al. osoitti AGPAT2: n tämän mutaation vaikuttamana entsyyminä2, 19. Tämän AGPAT2: n mutaation vuoksi triasyyliglyserolia ei synny lainkaan tai vain vähän muiden isoformien vaikutuksesta. AGPAT2 knockout-hiirten fenotyyppi on samanlainen kuin ihmisillä, joilla on CGL-tyyppi, Mikä vahvistaa tämän entsyymin roolin patofysiologiassa20, 21.
CGL Type 2. Magre ym. olivat ensimmäiset, jotka tunnistivat mutaation seipin-geenissä (kromosomi 11q13)3. Seipin-geenin (bscl2) mutaatiot (enimmäkseen hölynpölyä) tuottavat typistettyä proteiinia ja voivat vaikuttaa lipidimetaboliaan eri mekanismeilla: a) seipiinin stabiilisuuden väheneminen; B) lipiinin sitomiskyvyn väheneminen 1; ja C) epäonnistuminen oligomerisoitumisessa ja lokalisoitumisessa yksinomaan er membrane11: een. Jotkut solut pystyvät edelleen tuottamaan triasyyliglyserolia ja pieniä lipidipisaroita, mutta suuret lipidipisarat puuttuvat näiden pienten lipidipisaroiden fuusiokyvyn menetyksestä. Myös adipogeenisten tekijöiden, kuten peroksisomiproliferaattorilla aktivoidun Gamma-reseptorin (PPARG), sekä adiponektiinin ja adiposyyttien rasvahappoa sitovan proteiinin (FABP4)11, 16, ilmentymisessä on puutteita. Seipiinin puutos heikentää adipogeneesiä, lisää lipolyysiä ja estää triglyseridien kertymistä adiposyytteihin.
CGL tyyppi 3. Tämä tyyppi kuvattiin äskettäin potilaalla, jolla CGL-fenotyypistä huolimatta ei ollut mutaatioita geeneissä AGPAT2 tai bscl222. Hiiret, joilla on mutaatio cav1: ssä, ovat vastustuskykyisiä ruokavalion aiheuttamalle lihavuudelle ja niillä on insuliiniresistenssi, hypertriglyseridemia, vähentynyt adiponektiini, vähentynyt rasvamassa ja pienet adipocytes16. Valittuaan kandidaattigeenit hiirillä tehtyjen tutkimusten perusteella, Kim et al. vahvisti, että caveolin-1-geenissä (CAV1) on nonsense-mutaatio kromosomissa 7q3122.
CGL-tyyppi 4. Tässä on harvinainen tyyppi vaikuttaa geeni on CAVIN1, joka koodaa proteiinia Cavin-1. Ihmisillä sitä on raportoitu potilailla, joilla on yleistynyt synnynnäinen lipodystrofia ja lihasdystrofia15, 23.
hiljattain on kuvattu myös pcyt1a-ja PPARG-geenien mutaatioita, jotka aiheuttavat lipodystrofia24, 25.
kliiniset piirteet
CGL-potilailla esiintyy yleensä akromegaloidisia kasvoja, akantoosinegrikaaneja, febomegaliaa, hepatomegaliaa ja lihasten hypertrofia5, 26, 27. Useat kirjoittajat mainitsevat napatyrän oireyhtymän26 kliinisenä löydöksenä. Arvioimme sen esiintymistiheyttä potilassarjassamme, eikä yksikään heistä esittänyt tätä muutosta 28. Periumbilisen rasvakudoksen puuttuminen aiheuttaakin napanuoran pullistumisen, ja tämä saatetaan virheellisesti diagnosoida hernia28, 29-taudiksi.
kun adiposyytit eivät pysty varastoimaan rasvaa riittävästi, se kerääntyy muihin kudoksiin kuten maksaan ja lihaksiin aiheuttaen vaikean insuliiniresistenssin. Luun tiheysmittauksessa (DXA)voidaan havaita normaali tai korkea luun mineraalitiheys30 ja vähentää kehon kokonaisrasvaa (yleensä alle 6%) 27. Alhaisen kehon rasvan seurauksena seerumin adiponektiini ja leptiini ovat liian vähä27. Koska leptiini on välttämätön nälän hillitsemisessä, näillä potilailla on tyypillisesti ylensyöminen, joka on helposti havaittavissa lapsuudesta lähtien. Adiponektiinilla on tärkeä rooli insuliiniherkistäjänä, ja sen puute pahentaa insuliiniresistenssiä. Tästä huolimatta aluksi glukoosi ja glykoitunut hemoglobiini ovat normaaleja hyvin korkeiden insuliinitasojen kustannuksella. Diabetes alkaa yleensä murrosiässä; meidän sarjoissamme puhkeamisiän keskiarvo oli 15,8±7,1 vuotta27. Aluksi niitä ohjataan suun kautta otettavilla lääkkeillä, jotka tarvitsevat suuria insuliiniannoksia muutamassa vuosissa27. Arteriaalinen hypertensio esiintyy kolmanneksella potilaista27.
kussakin CGL-tyypissä on joitakin erityisiä kliinisiä piirteitä. Tyypin 1 potilailla esiintyy edelleen mekaanista rasvakudosta, erityisesti kämmenissä, jalkapohjissa, orbitaaleissa, peri-nivelalueissa31. Sen sijaan tyypin 2 potilailla ei ole metabolisia eikä mekaanisia rasvakudoksia. Seipiiniä esiintyy voimakkaasti aivoissa ja pikkuaivoissa ja se osallistuu myös hermotoimintojen säätelyyn. Yli puolella tyypin 2 potilaista on jonkin verran kognitiivisia heikentymiä1, 8. Tyypeillä 3 ja 4 on mekaanisen ja luuytimen rasvan säilyminen, ja tyypillä 4 on lihasheikkous, joka liittyy korkeaan seerumin kreatiinikinaasiin ja selkärangan epävakauteen15.
on myös sukupuolikohtaisia kliinisiä piirteitä. Munasarjojen monirakkulatauti ja kuukautisia ovat yleisiä32. Kuukautiskierto palautuu yleensä normaaliksi metreleptiinin käytön myötä, mikä johtuu todennäköisesti insuliiniherkkyyden paranemisesta ja LH-pulsatiliteetin palautumisesta32. Tyypin 2 miehillä voi olla teratozoospermia, koska sukusoluissa ei ole seipiiniä 33.
hypertriglyseridemiaa esiintyy ensimmäisten elinvuosien jälkeen ja se voi aiheuttaa akuutin haimatulehduksen. HDL on yleensä alle 30 mg / dL. Maksaentsyymien nousu on myös varhainen löydös ja tulevat rasvan kertymisestä maksaan. Seerumin trombosyyttiarvojen asteittainen lasku viittaa maksasairauden pahenemiseen ja todennäköiseen kirroosiin34.
koska Cavin-1 esiintyy lihassoluissa, tyypin 4 potilailla on lievä lihasheikkous ja kohonnut kreatiinikinase15.
elinajanodote, lähinnä tyypin 2 kohdalla, on huomattavasti lyhentynyt, eikä kuolema tapahdu harvoin ennen 30 vuoden ikää (henkilökohtainen kokemus perustuu 20 potilaaseen, jotka ovat kuolleet viimeisten 19 vuoden aikana). Kuolinsyyt liittyvät diabetekseen (munuaisten vajaatoiminta, äkkikuolema), maksaan (kirroosi, ruoansulatuskanavan verenvuoto) tai infektioihin.
diagnoosi ja hoito
CGL-diagnoosi perustuu kliiniseen tietoon: akromegaloidipiirteet, akantoosi nigrikaanit, kehon kokonaisrasvan väheneminen, lihasten hypertrofia ja napanuoran ulkonema. Myös laboratoriotiedot voivat osoittaa diabetesta, jolla on vaikea insuliiniresistenssi ja hypertriglyseridemia. Kuvantamistestit voivat auttaa tunnistamaan kohdunulkoinen talletuksia rasvaa lähinnä maksassa ja haimassa (maksan rasvoittuminen hepatomegalia ja haiman rasva). DXA voi vahvistaa alhaisen kehon rasvan ja korkean luun tiheyden30.
CGL: n hoito koostuu ruokavalion tiukasta valvonnasta ja rasvan, pääasiassa triglyseridien ja korkean glykeemisen indeksin omaavien elintarvikkeiden saannin vähentämisestä liitännäissairauksien ehkäisemiseksi ja hallitsemiseksi 29. Ihanneruokavalio on kuitenkin haastava tavoite, koska ruokahalu on lisääntynyt ja sitä suositaan ankarasti. Liikuntaa on myös kannustettava parantamaan oheissairauksien hallintaa, lukuun ottamatta potilaita, joilla on vasta-aiheita, kuten vaikea kardiomyopatia29.
lääkehoidon osalta näitä potilaita voidaan hoitaa tavanomaisilla diabetes -, verenpainetauti-ja dyslipidemialääkkeillä. Ensimmäinen vaihtoehto diabeteksen ja insuliiniresistenssin hoitoon on metformiini, mutta yleensä se ei riitä. Toisin kuin osittaisen lipodystrofian hoidossa, tiatsolidiinidioneja tulee käyttää caution29: n kanssa. Muitakin suun kautta otettavia diabeteslääkkeitä käytetään, mutta niitä ei ole erityisesti tutkittu CGL-potilailla. Eläimillä on tietoja, jotka viittaavat siihen, että SGLT2-inhibiittorien (dapagliflotsiini) käytöllä voi olla etuja kardiomyopatian ehkäisemisessä35; tutkimukset ovat tarpeen tämän vahvistamiseksi ihmisillä. Sairauden edetessä ja vaikean insuliiniresistenssin ilmetessä tarvitaan suuria vuorokausiannoksia. Ihonalaisen rasvakudoksen puute on ongelma annettaessa suuria insuliiniannoksia. Väkevämpää insuliinia (U-300 tai U500) voidaan tarvita 36. Näillä potilailla esiintyy vakava dyslipidemia, joka johtuu pääasiassa triglyseridien noususta ja alhaisesta HDL: stä, ja siksi fibraatin käyttö on joskus tarpeen akuutin haimatulehduksen estämiseksi. Lisäksi näiden potilaiden suuren sydän-ja verisuonitautiriskin vuoksi statiinihoitoa tulee harkita ja LDL: n tai ei-HDL: n tavoitearvon tulee olla strict29.
päivittäiset metreleptiinipistokset vähentävät merkitsevästi ruokahalua ja tuovat hyötyjä alentamalla glykemiaa, triglyseridemiaa ja maksaentsyymejä. Erityisesti lapsilla on huomattavaa vatsan ympärysmitan pieneneminen, mikä johtuu todennäköisesti hepatomegalian vähenemisestä.
Conclusion
CGL on harvinainen ja vaikea sairaus, joka voi ilmetä diabeteksen yhteydessä (vaatii yleensä suuria insuliiniannoksia) ja ennenaikaisena kuolemana. Potilaan fenotyyppi on varsin tyypillinen, mikä edellyttää kuitenkin terveydenhuollon ammattilaisten tietämystä oireyhtymästä varhaisen diagnoosin tekemiseksi. Metreleptiini näyttää olevan tällä hetkellä ainoa lääke, joka voi muuttaa taudin luonnollista historiaa.
eturistiriita: ei ole.
- Nolis T. tutkii yleisempien geneettisten ja hankittujen lipodystrofioiden taustalla olevaa patofysiologiaa. Journal of human genetics. 2014 tammi; 59 (1): 16-23.
- Agarwal AK, Arioglu E, De Almeida s, et al. AGPAT2 mutatoituu synnynnäisessä yleistyneessä lipodystrofiassa, joka liittyy kromosomiin 9q34. Nat Genet. 2002 toukokuu; 31(1): 21-3.
- Magre J, Delepine M, Khallouf E, et al. Synnynnäisessä Berardinelli-Seip-lipodystrofiassa muutetun geenin tunnistaminen kromosomissa 11q13. Luonnongenetiikka. 2001 Aug; 28 (4): 365-70.
- Patni N, Garg A. Synnynnäiset yleistyneet lipodystrofiat — uusia oivalluksia aineenvaihdunnan häiriöistä. Nature reviews Endokrinologia. 2015 syys; 11 (9): 522-34.
- Garg A. hankitut ja perityt lipodystrofiat. New England journal of medicine. 2004 Mar 18; 350 (12): 1220-34.
- de Azevedo Medeiros LB, Candido Dantas VK, Craveiro Sarmento AS, et al. Berardinelli-Seipin synnynnäisen lipodystrofian suuri esiintyvyys Rio Grande do Norten osavaltiossa Koillis-Brasiliassa. Diabetol Metab Syndr. 2017; 9: 80.
- Chiquette E, Oral EA, Garg A, et al. Yleisen ja osittaisen lipodystrofian esiintyvyyden arviointi: löydökset ja haasteet. Diabetes, metabolinen oireyhtymä ja lihavuus: tavoitteet ja hoito. 2017: 375-83.
- Wee K, Yang W, Sugii s, et al. Lipodystrofian ja seipiinin toimintojen mekanistiseen ymmärtämiseen. Biotieteiden raportit. 2014; 34(5).
- Dollet L, Magre J, Cariou B, et al. Toiminto seipin: uusia oivalluksia bscl2 / seipin knockout hiiri malleja. Biochimie. 2014 tammi; 96: 166-72.
- Sim MF, Dennis RJ, Aubry EM, et al. Ihmisen lipodystrofiaproteiini seipiini on ADIPOGEENISEN PA-fosfataasi lipiini 1: n ER-kalvosovitin. Molekyylimetaboliaa. 2012; 2(1): 38-46.
- Sim MF, Talukder MM, Dennis RJ, et al. Ihmisen lipodystrofiaproteiini seipinin luonnossa esiintyvien mutaatioiden analyysi paljastaa useita mahdollisia patogeenisiä mekanismeja. Diabetologia. 2013 marras; 56 (11): 2498-506.
- Payne VA, Grimsey N, Tuthill A, et al. Ihmisen lipodystrofiageeni BSCL2/seipiini saattaa olla välttämätön rasvasolujen normaalin erilaistumisen kannalta. Diabetes. 2008 Aug; 57 (8): 2055-60.
- Cohen AW, Hnasko R, Schubert W, et al. Caveolae ja caveolins rooli terveyden ja sairauden. Fysiologiset arviot. 2004 loka; 84 (4): 1341-79.
- Pilch PF, Liu L. Fat caveolae: caveolae, lipidikauppa ja rasva-aineenvaihdunta adiposyyteissä. Endokrinologian ja aineenvaihdunnan suuntaukset: TEM. 2011 Aug; 22 (8): 318-24.
- Hayashi YK, Matsuda C, Ogawa M, et al. Ihmisen PTRF-mutaatiot aiheuttavat toissijaista kaveoliinien puutosta, joka johtaa lihasdystrofiaan, johon liittyy yleistynyt lipodystrofia. J Clin Invest. 2009 syys; 119 (9): 2623-33.
- Parton RG, del Pozo MA. Caveolae plasmakalvon antureina, suojina ja järjestäjinä. Nature tutkii Molekyylisolubiologiaa. 2013 helmi; 14 (2): 98-112.
- Le Lay S, Briand N, Blouin CM, et al. Lipoatrofinen caveolin – 1-puutteellinen hiirimalli paljastaa autofagian kypsissä adiposyyteissä. Autofagiaa. 2010 Aug; 6(6): 754-63.
- Liu L, Brown D, McKee M, et al. Cavin/PTRF: n poisto aiheuttaa maailmanlaajuista kaveolae -, dyslipidemia-ja glukoosi-intoleranssin menetystä. Solujen aineenvaihdunta. 2008 loka; 8 (4): 310-7.
- Garg A, Wilson R, Barnes R, et al. Synnynnäinen yleistynyt lipodystrofia-geeni kartoittaa ihmisen kromosomiin 9q34. Journal of clinical endocrinology and metabolism. 1999 syys; 84 (9): 3390-4.
- Vogel P, Read R, Hansen G, et al. Synnynnäisen yleistyneen lipodystrofian patologia Agpat2 – / – hiirillä. Eläinlääketieteellinen patologia. 2011 toukokuu; 48(3): 642-54.
- Cortes VA, Curtis DE, Sukumaran s, et al. Maksan rasvoittumisen molekyylimekanismit ja insuliiniresistenssi synnynnäisen yleistyneen lipodystrofian agpat2-puutteellisessa hiirimallissa. Solujen aineenvaihdunta. 2009 helmi; 9 (2): 165-76.
- Kim CA, Delepine M, Boutet E, et al. Homotsygoottisen nonsense caveolin-1-mutaation ja Berardinelli-Seip: n synnynnäisen lipodystrofian välinen yhteys. J Clin Endocrinol Metab. 2008 huhti; 93 (4): 1129-34.
- Rajab A, Straub V, McCann LJ, et al. Kuolemaan johtava sydämen rytmihäiriö ja pitkän QT-ajan oireyhtymä uudessa synnynnäisessä yleistyneessä lipodystrofiassa, johon liittyy ptrf-CAVIN-mutaatioiden aiheuttama lihasreppling (CGL4). PLOS genetics. 2010 12. Maaliskuuta; 6(3): e1000874.
- Payne F, Lim K, Girousse A, et al. Kennedyn fosfatidyylikoliinireittiä häiritsevät mutaatiot synnynnäisestä lipodystrofiasta ja rasvamaksasta kärsivillä ihmisillä. Proc Natl Acad Sci U S A. 2014 Jun 17; 111 (24): 8901-6.
- Dyment DA, Gibson WT, Huang L, et al. PPARG: n bialleliset mutaatiot aiheuttavat synnynnäisen, yleistyneen lipodystrofian, joka muistuttaa Berardinelli-Seip-oireyhtymää. EUR J Med Genet. 2014 syys; 57 (9): 524-6.
- Garg A. Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. Journal of clinical endocrinology and metabolism. 2011 marras; 96 (11): 3313-25.
- Lima JG, Nobrega LH, de Lima NN, et al. Kliiniset ja laboratoriotiedot useista potilaista, joilla on synnynnäinen yleistynyt lipodystrofia. Diabetol Metab Syndr. 2016; 8: 23.
- Lima GJ, Lima NN, Oliveira CF, et al. Napatyrä potilailla, joilla on Berardinelliseip-oireyhtymä: Onko se todella tyrä. J Clin Mol Endocrinol. 2015; 1(1): 3.
- Brown RJ, Araujo-Vilar D, Cheung PT, et al. Lipodystrofia Syndromes: a Multi-Society Practice Guideline. J Clin Endocrinol Metab. 2016 joulu; 101 (12): 4500-11.
- Lima JG, Nobrega LH, Lima NN, et al. Synnynnäistä Berardinelli-Seip-lipodystrofiaa sairastavilla potilailla luun tiheys on suurempi Trabekkelikohdissa ja tyypin 2 potilailla. J Clin Densitom. 2016 marras 25.
- Simha V, Garg A. fenotyyppinen heterogeenisuus kehon rasvan jakautumisessa potilailla, joilla on synnynnäinen yleistynyt lipodystrofia, joka johtuu AGPAT2-tai SEIPIN-geenien mutaatioista. J Clin Endocrinol Metab. 2003 marras; 88 (11): 5433-7.
- Musso C, Cochran E, Javor E, et al. Rekombinantti metionyyli-ihmisen leptiinihoidon pitkäaikainen vaikutus hyperandrogenismiin ja kuukautistoimintoihin naisilla ja aivolisäkkeellä hypoleptineemisillä lipodystrofiapotilailla sekä miehillä että naisilla. Aineenvaihdunta. 2005 helmi; 54 (2): 255-63.
- Jiang M, Gao M, Wu C, et al. Kivesten seipiinin puute aiheuttaa miehillä teratozoospermia-oireyhtymää. Proc Natl Acad Sci U S A. 2014 May 13; 111 (19): 7054-9.
- Mitchell O, Feldman DM, Diakow M, et al. Trombosytopenian patofysiologia kroonisessa maksasairaudessa. Hepat Med. 2016; 8: 39-50.
- Joubert M, Jagu B, Montaigne D, et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes. 2017 Apr; 66(4): 1030-40.
- Lima JG, Lima NN, Lima RLM, et al. Glargine U300 Insulin as a Better Option than Degludec U100 to Treat a Congenital Generalized Lipodystrophy Patient. Clin Diabetes Res. 2017; 1(1).