Article
Josivan Gomes Lima1 *, Marcel Catão Ferreira dos Santos1, Julliane Tamara Araújo de Melo Campos2
1Départemento de medicina clínica, disciplina de endocrinologia e metabologia. Hospital Universitário Onofre Lopes, Universidade Federal do Rio Grande do Norte (UFRN), Natal, RN, Brésil
2Faculté des Sciences de la santé de Trairi, Université Fédérale du Rio Grande do Nord (UFRN), Natal, RN, Brésil
Résumé
La Lipodystrophie généralisée congénitale (CGL) est une maladie autosomique récessive rare et sévère maladie. Les patients sont défectueux dans le stockage de la graisse corporelle et, par conséquent, ils déposent de la graisse dans les tissus ectopiques, principalement le foie, et peuvent développer une cirrhose. La résistance à l’insuline est une découverte typique, provoquant un diabète qui nécessite des doses quotidiennes élevées d’insuline. Dans l’État du Rio Grande do Norte, au Brésil, nous avons l’une des plus grandes cohortes de patients atteints de LMC. Dans cet article, nous passons en revue la physiopathologie, le tableau clinique et le traitement de cette maladie.
Introduction
Le diabète de type 2 est un problème de santé mondial et résulte généralement d’un poids excessif et d’une augmentation de la graisse viscérale entraînant une résistance à l’insuline périphérique et une incapacité du pancréas à libérer de l’insuline pour compenser cette résistance. D’autres types de diabète moins courants sont dus à des mutations génétiques spécifiques, comme la Lipodystrophie congénitale généralisée (CGL), également connue sous le nom de Lipodystrophie congénitale Berardinelli-Seip (BSCL). CGL est une maladie autosomique récessive qui est classée en quatre types, en fonction de la mutation génétique. Les gènes altérés jouent des fonctions essentielles pour la formation des adipocytes, la production de lipides et le stockage approprié à l’intérieur de l’adipocyte. Les mutations diminuent le tissu adipeux avec pour conséquence le dépôt de graisse dans les sites ectopiques, provoquant un foie gras, une altération du métabolisme des glucides, une résistance à l’insuline sévère avec hyperinsulinémie et caractéristiques acromégaloïdes et une dyslipidémie1-3. Le syndrome de CGL a environ 500 cas signalés dans le monde. Au Brésil, dans l’État du Rio Grande do Norte (RN), nous avons diagnostiqué, traité et suivi 54 cas au cours des 20 dernières années4,5. Dans une étude descriptive utilisant des données secondaires, nous avons estimé un total de 103 patients sous RN6. Cela indique une prévalence beaucoup plus élevée que celle rapportée dans la littérature (1: 1 million) 7.
Formation et stockage de triacylglycérol dans des gouttelettes lipidiques
La biosynthèse des triglycérides et des phospholipides (Figure 1A) commence par la glycérol-3-phosphate acyltransférase (GPAT) acylant le glycérol-3-phosphate en position 1, formant le 1-acylglycérol-3-phosphate (acide lysophosphatidique). Elle est suivie d’une autre étape d’acylation en position deux par l’enzyme AGPAT (1-Acylglycérol-3-phosphate acyltransférase), à l’origine du 1,2-Diacylglycérol-3-phosphate (acide phosphatidique). C’est une étape intermédiaire clé dans la voie de biosynthèse des triglycérides et des phosphoglycérides. Il existe 11 isoformes d’enzymes AGPAT, codées par différents génes4. AGPAT1 et AGPAT2 sont les plus étudiés. AGPAT1 est présent à des niveaux élevés dans les testicules, le pancréas et, dans une moindre mesure, dans le tissu adipeux et d’autres tissus comme le cœur, le placenta, le cerveau et les poumons, tandis qu’AGPAT2 est abondant dans les tissus adipeux. Dans les étapes suivantes, l’enzyme cytosolique phosphatidique phosphatase de l’acide (PAP ou lipine) est à l’origine du 1,2-diacylglycérol et la 1,2-diacylglycérol acyltransférase (DGAT) forme du triacylglycérol4. L’acide phosphatidique et le diacylglycérol peuvent également être à l’origine d’autres phospholipides tels que la cardiolipine, le phosphatidylinositol et la phosphatidylcholine.
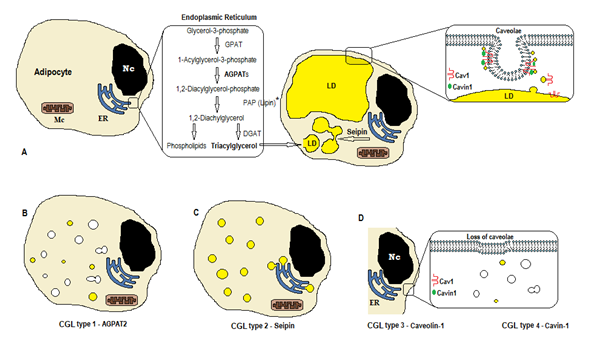
Figure 1. Schéma de synthèse des triglycérides selon les types CGL. (A) Synthèse et stockage normaux du triacylglycérol (TAG) dans l’adipocyte. (B) La mutation d’AGPAT2 diminue la production d’ÉTIQUETTES (certaines sont encore synthétisées sous la stimulation d’autres AGPAT). (C) Mutation de la synthèse des étiquettes de diminution du gène seipin et formation et fusion de gouttelettes lipidiques (LD). (D) La cavéoline-1 et la cavine-1 sont nécessaires à la formation et à la stabilisation des cavéoles. La mutation de CAV1 (type 3) ou de CAVIN1 (type 4) peut entraîner une perte de cavéoles dans la membrane. Nc, noyau. Réticulum endoplasmique. Mc, mitochondries. * La lipine est une enzyme cytosolique ancrée par la seipine dans les URGENCES.
Ces réactions se produisent dans le réticulum endoplasmique (RE) des adipocytes, où une accumulation progressive de triglycérides provoque la formation de petites gouttelettes lipidiques (LD)8. Le produit du gène BSCL2 est une protéine transmembranaire appelée seipine qui provoque la fusion de petites LD, à l’origine de grandes LD. La seipine réside dans l’ER et se concentre à la jonction avec la LD naissante, facilitant le trafic lipidique entre l’ER et la LD et l’incorporation de triglycérides dans la LD9. La seipine peut également servir d’ancrage ER à l’enzyme cytosolique lipine 1. En plus d’être nécessaire à la fusion, à la taille et à la morphologie des gouttelettes lipidiques, la seipine est également essentielle à l’adipogenèse (via l’interaction avec la lipine 1) et à la lipolyse des triglycérides cellulaires10,11. Une carence en seipine entrave la différenciation des pré-adipocytes en adipocytes et affecte la maturation finale9, comme l’ont montré des études sur des cellules souches mésenchymateuses avec BSCL2 assommée12. Les tissus non adipeux expriment également la seipine et d’autres fonctions doivent être déterminées.
Dans les adipocytes, les cavéoles, qui sont des invaginations membranaires spécialisées de 50 à 100 nm, représentent 20% de la surface de la membrane plasmique, faisant des adipocytes les cellules ayant la plus forte densité de cavéoles13. La formation de gouttelettes lipidiques nécessite une protéine membranaire (Cavéoline – le composant principal des membranes des cavéoles) et une protéine cytoplasmique (Cavine-1) 14. Les gènes CAV1, CAV2 et CAV3 codent trois formes de cavéoline ayant des structures similaires (Cavéoline-1, Cavéoline-2 et Cavéoline-3, respectivement). La cavéoline-1 et la cavéoline-2 sont présentes dans les adipocytes, les fibroblastes et les cellules endothéliales, et la cavéoline-3 n’est présente que dans le muscle squelettique et cardiaque13,15. La cavéoline-1 est la plus importante et la plus étudiée. Il est exprimé en deux isoformes différentes (1a et 1b). La cavéoline-1 se transfère de la membrane plasmique en gouttelettes lipidiques, ce qui est nécessaire au trafic et au métabolisme16 des lipides. Les gouttelettes lipidiques stockent les triglycérides après l’alimentation et ces molécules sont hydrolysées en acide gras et libérées pendant le jeûne; ce mécanisme peut être régulé par la cavéoline-116. Le déficit en cavéoline-1 augmente également la susceptibilité à la mort cellulaire par autophagie17.
Le gène CAVIN1 code pour une protéine cytoplasmique appelée caveolae associated protein 1 (Cavin-1) 14, 16, qui est obligatoire pour la formation et la stabilisation des caveolae. La cavine-1 est exprimée dans les adipocytes, les cellules musculaires et d’autres cellules et est également essentielle à la transmission des signaux d’origine des cavéoles14,18. Le knockout du gène CAV1 provoque un manque de cavéoles dans les cellules non musculaires, tandis que le knockout de CAVIN1 provoque l’absence de cavéoles dans tous les tissus, y compris le muscle14. L’absence de cavéoles peut affecter la régulation de la lipolyse, du flux d’acides gras, de la synthèse des triglycérides et des signaux d’autres voies.
Types de CGL
Sur la base d’altérations génétiques détectables, quatre types sont décrits. Les types 1 et 2 sont responsables de plus de 95% des cas, et le type 2 a un phénotype plus gravement affecté. Un seul cas de type 3 et environ 30 cas de type 4 ont été signalés4.
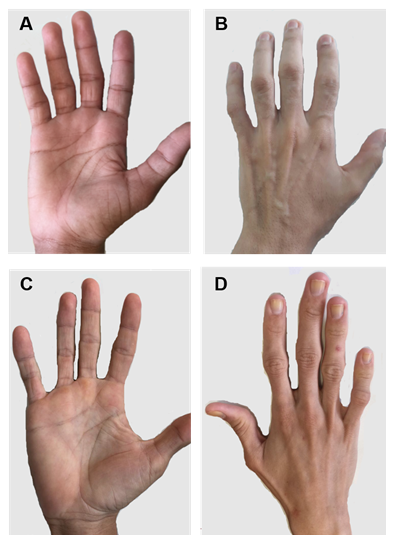
Figure 2. Mains de patients atteints de CGL de types 1 et 2. (A) et (B) Vues antérieures et postérieures des mains des patients de type 1. Mains apparemment normales, car il y a encore du tissu adipeux mécanique. (C) et (D) Vues antérieures et postérieures des mains des patients de type 2. La gravité de la maladie est plus grande et le manque de graisse est évident et facilement perceptible.
CGL Type 1. En 1999, Garg et coll. décrit la mutation des patients sur le chromosome 9q34, et trois ans plus tard Agarwal et al. a montré AGPAT2 comme l’enzyme affectée par cette mutation2,19. En raison de la mutation de cet AGPAT2, aucune ou une production minimale de triacylglycérol ne se produit par le stimulus d’autres isoformes. Le phénotype des souris knockout AGPAT2 est similaire à celui des humains de type CGL, confirmant le rôle de cette enzyme dans la physiopathologie20,21.
CGL Type 2. Magre et coll. ont été les premiers à identifier la mutation du gène seipin (chromosome 11q13) 3. Les mutations (principalement non-sens) du gène de la seipine (BSCL2) produisent une protéine tronquée et peuvent affecter le métabolisme des lipides par différents mécanismes: a) diminution de la stabilité de la seipine; b) réduction de la capacité de se lier à la lipine 1; et c) incapacité à s’oligomériser et à se localiser exclusivement sur la membrane er11. Certaines cellules sont encore capables de générer du triacylglycérol et de petites gouttelettes lipidiques, mais les grosses gouttelettes lipidiques sont absentes en raison de la perte de la capacité de fusion de ces petites gouttelettes lipidiques. Il existe également une défaillance dans l’expression de facteurs adipogènes, tels que le récepteur gamma activé par le proliférateur du peroxysome (PPARG), ainsi que l’adiponectine et la protéine de liaison aux acides gras adipocytaires (FABP4) 11, 16. La carence en seipine altère l’adipogenèse, augmente la lipolyse et empêche l’accumulation de triglycérides dans les adipocytes.
CGL type 3. Ce type a été décrit récemment chez un patient qui, malgré son phénotype CGL, n’avait pas de mutations dans les gènes AGPAT2 ou BSCL222. Les souris avec une mutation de Cav1 sont résistantes à l’obésité induite par le régime alimentaire et présentent une résistance à l’insuline, une hypertriglycéridémie, une diminution de l’adiponectine, une réduction de la masse grasse et de petites adipocytes16. Après avoir choisi des gènes candidats sur la base d’études chez la souris, Kim et al. a confirmé la présence d’une mutation non-sens dans le gène caveolin-1 (CAV1), sur le chromosome 7q3122.
CGL type 4. Dans ce type rare, le gène affecté est le CAVIN1, qui code pour la protéine Cavin-1. Chez l’homme, elle a été rapportée chez des patients atteints de lipodystrophie congénitale généralisée et de dystrophie musculaire15,23.
Récemment, des mutations dans les gènes PCYT1A et PPARG ont également été décrites provoquant une lipodystrophie 24,25.
Caractéristiques cliniques
Les patients atteints de LMC présentent habituellement un faciès acromégaloïde, une acanthose nigricane, une phébomégalie, une hépatomégalie et une hypertrophie musculaire5, 26, 27. Plusieurs auteurs citent la hernie ombilicale comme découverte clinique du syndrome26. Nous en avons évalué la fréquence dans notre série de patients, et aucun d’entre eux n’a présenté ce changement28. En fait, l’absence de tissu adipeux périumbilical provoque une protrusion de la cicatrice ombilicale, ce qui peut être diagnostiqué à tort comme une hernie28,29.
Une fois que les adipocytes ne peuvent pas stocker suffisamment de graisse, ils s’accumulent dans d’autres tissus tels que le foie et les muscles, provoquant une résistance sévère à l’insuline. La densitométrie osseuse (DXA) peut montrer une densité minérale osseuse normale ou élevée 30 et une réduction de la graisse corporelle totale (généralement inférieure à 6%)27. En raison d’une faible teneur en graisse corporelle, l’adiponectine sérique et la leptine sont trop faibles27. Comme la leptine est essentielle pour contrôler la faim, ces patients souffrent généralement d’hyperphagie, ce qui est facilement apparent depuis l’enfance. L’adiponectine joue un rôle important en tant que sensibilisant à l’insuline et son absence aggrave la résistance à l’insuline. Malgré cela, au départ, le glucose et l’hémoglobine glyquée sont normaux au détriment de taux d’insuline très élevés. Le diabète commence habituellement à la puberté; dans notre série, l’âge moyen d’apparition était de 15,8 ±7,1 ans27. Initialement, ils sont contrôlés par des médicaments oraux, nécessitant de fortes doses d’insuline en quelques années27. L’hypertension artérielle survient chez un tiers des patients27.
Il existe certaines caractéristiques cliniques spécifiques de chaque type de LMC. Les patients de type 1 présentent toujours de la graisse adipeuse mécanique, en particulier dans les paumes, les semelles, les régions orbitales et périarticulaires 31. En revanche, les patients de type 2 montrent une absence de tissus adipeux métaboliques et mécaniques. Seipin est fortement exprimé dans le cerveau et le cervelet et est également impliqué dans la régulation des fonctions neuronales. Plus de la moitié des patients de type 2 présentent des troubles cognitifs1,8. Les types 3 et 4 ont une préservation de la graisse mécanique et de la moelle osseuse, et le type 4 présente une faiblesse musculaire associée à une créatine kinase sérique élevée et à une instabilité spinale15.
Il existe également des caractéristiques cliniques spécifiques au sexe. Les ovaires polykystiques et l’aménorrhée sont fréquents32. Les cycles menstruels reviennent généralement à la normale avec l’utilisation de la métréleptine, probablement en raison de l’amélioration de la sensibilité à l’insuline et de la restauration de la pulsatilité de la LH32. Les hommes de type 2 peuvent avoir une tératozoospermie en raison de l’absence de seipine dans les cellules germinales33.
L’hypertriglycéridémie survient dès les premières années de la vie et peut provoquer une pancréatite aiguë. Le HDL est généralement inférieur à 30 mg / dL. L’élévation des enzymes hépatiques est également une découverte précoce et provient du dépôt de graisse dans le foie. Des réductions progressives des plaquettes sériques suggèrent une aggravation de la maladie du foie et une cirrhose probable34.
Comme la Cavine-1 est présente dans les cellules musculaires, les patients de type 4 présentent une faiblesse musculaire légère et une augmentation de la créatine kinase15.
L’espérance de vie, principalement dans le type 2, est considérablement réduite, le décès survenant rarement avant l’âge de 30 ans (expérience personnelle basée sur 20 patients décédés au cours des 19 dernières années). Les causes de décès sont liées au diabète (insuffisance rénale, mort subite), au foie (cirrhose, saignements digestifs) ou aux infections.
Diagnostic et traitement
Le diagnostic de CGL est basé sur des données cliniques: caractéristiques acromégaloïdes, acanthose nigricane, réduction de la graisse corporelle totale, hypertrophie musculaire et protrusion de la cicatrice ombilicale. En outre, les données de laboratoire peuvent montrer un diabète avec une résistance à l’insuline sévère et une hypertriglycéridémie. Les tests d’imagerie peuvent aider à identifier les dépôts ectopiques de graisse principalement dans le foie et le pancréas (stéatose hépatique avec hépatomégalie et stéatose pancréatique). Le DXA peut confirmer le faible taux de graisse corporelle et la densité osseuse élevée30.
Le traitement du CGL consiste en un contrôle strict de l’alimentation avec diminution de l’apport en matières grasses, principalement des triglycérides et des aliments à indice glycémique élevé pour prévenir et contrôler les comorbidités29. Cependant, le régime idéal est un objectif difficile à atteindre en raison de l’augmentation de l’appétit et de la restriction sévère préconisée. L’activité physique doit également être encouragée pour améliorer le contrôle des comorbidités, sauf chez les patients présentant des contre-indications telles que la cardiomyopathie grave29.
En ce qui concerne le traitement médicamenteux, ces patients peuvent être traités avec les médicaments habituels pour le diabète, l’hypertension et la dyslipidémie. Le premier choix pour le traitement du diabète et de la résistance à l’insuline est la metformine, mais généralement, cela ne suffit pas. Contrairement au traitement de la lipodystrophie partielle, les thiazolidinediones doivent être utilisées avec prudence29. D’autres antidiabétiques oraux sont utilisés, mais ils n’ont pas été spécifiquement étudiés chez les patients atteints de LMC. Des données sur des animaux suggèrent que l’utilisation d’inhibiteurs de la SGLT2 (dapagliflozine) pourrait avoir des avantages à prévenir la cardiomyopathie35; des études sont nécessaires pour le confirmer chez l’homme. À mesure que la maladie progresse et que la résistance à l’insuline sévère se produit, des doses quotidiennes élevées d’insuline sont nécessaires. Le manque de tissu adipeux sous-cutané est un problème lors de l’administration des fortes doses d’insuline. Une insuline plus concentrée (U-300 ou U500) peut être nécessaire36. Ces patients présentent une dyslipidémie sévère, principalement due à l’augmentation des triglycérides et à une faible teneur en HDL, et par conséquent, l’utilisation de fibrate est parfois nécessaire pour prévenir la pancréatite aiguë. De plus, en raison du risque cardiovasculaire élevé de ces patients, une intervention avec une statine doit être envisagée et les cibles de LDL ou de non-HDL doivent être strictes29.
Les injections quotidiennes de métréleptine provoquent une diminution significative de l’appétit et apportent des avantages en réduisant la glycémie, la triglycéridémie et les enzymes hépatiques. Il est notable, en particulier chez les enfants, la réduction de la circonférence abdominale, probablement due à une réduction de l’hépatomégalie.
Conclusion
La LPC est une maladie rare et grave qui peut survenir avec le diabète (nécessitant généralement de fortes doses d’insuline) et une mort précoce. Le phénotype du patient est assez caractéristique, nécessitant cependant une connaissance du syndrome par les professionnels de la santé pour établir un diagnostic précoce. La métréleptine semble être le seul médicament à l’heure actuelle capable de modifier l’histoire naturelle de la maladie.
Conflit d’intérêts : aucun.
- Nolis T. Explorant la physiopathologie derrière les lipodystrophies génétiques et acquises les plus courantes. Journal de génétique humaine. 2014 Jan; 59(1): 16-23.
- Agarwal AK, Arioglu E, De Almeida S, et al. AGPAT2 est muté dans une lipodystrophie généralisée congénitale liée au chromosome 9q34. Nat Genet. 2002 Mai; 31(1): 21-3.
- Magre J, Delépine M, Khallouf E, et al. Identification du gène altéré dans la lipodystrophie congénitale Berardinelli-Seip sur le chromosome 11q13. Génétique de la nature. 2001 Août; 28(4): 365-70.
- Patni N, Garg A. Lipodystrophies congénitales généraliséesnew de nouvelles perspectives sur le dysfonctionnement métabolique. La nature examine l’endocrinologie. 2015 Sept; 11(9): 522-34.
- Garg A. lipodystrophies acquises et héritées. Le journal de médecine de la Nouvelle-Angleterre. 18 mars 2004; 350(12): 1220-34.
- de Azevedo Medeiros LB, Candido Dantas VK, Craveiro Sarmento AS, et al. Prévalence élevée de la Lipodystrophie congénitale Berardinelli-Seip dans l’État du Rio Grande do Norte, au nord-est du Brésil. Diabétol Metab Syndr. 2017; 9: 80.
- Chiquette E, Oral EA, Garg A, et al. Estimation de la prévalence de la lipodystrophie généralisée et partielle: résultats et défis. Diabète, Syndrome métabolique et Obésité: Cibles et thérapie. 2017: 375-83.
- Wee K, Yang W, Sugii S, et al. Vers une compréhension mécaniste de la lipodystrophie et des fonctions de la seipine. Rapports bioscientifiques. 2014; 34(5).
- Dollet L, Magre J, Cariou B, et al. Fonction de seipin: nouvelles perspectives des modèles de souris knockout Bscl2 / seipin. Biochimie. 2014 Jan; 96:166-72.
- Sim MF, Dennis RJ, Aubry EM, et al. La protéine de lipodystrophie humaine seipin est un adaptateur de membrane ER pour la lipine 1 de la PA phosphatase adipogène. Métabolisme moléculaire. 2012; 2(1): 38-46.
- Sim MF, Talukder MM, Dennis RJ, et al. L’analyse des mutations naturelles de la protéine de lipodystrophie humaine seipin révèle de multiples mécanismes pathogènes potentiels. Diabétologie. 2013 Novembre; 56 (11): 2498-506.
- Payne VA, Grimsey N, Tuthill A, et al. Le gène de la lipodystrophie humaine BSCL2 / seipin peut être essentiel à la différenciation normale des adipocytes. Diabète. 2008 Août; 57 (8): 2055-60.
- Cohen AW, Hnasko R, Schubert W, et al. Rôle des cavéoles et des cavéolines dans la santé et la maladie. Examens physiologiques. 2004 Octobre; 84(4): 1341-79.
- Pilch PF, Liu L. Grottes de graisse: cavéoles, trafic de lipides et métabolisme des lipides dans les adipocytes. Tendances en endocrinologie et métabolisme: TEM. 2011 Août; 22 (8): 318-24.
- Hayashi YK, Matsuda C, Ogawa M, et al. Les mutations PTRF humaines provoquent une carence secondaire en cavéolines entraînant une dystrophie musculaire avec lipodystrophie généralisée. J Clin Invest. 2009 Sep; 119(9): 2623-33.
- Parton RG, del Pozo MA. Caveolae comme capteurs, protecteurs et organisateurs de membranes plasmiques. Nature examine la biologie cellulaire moléculaire. 2013 Fév; 14 (2): 98-112.
- Le Lay S, Briand N, Blouin CM, et al. Le modèle de souris déficiente en cavéoline-1 lipoatrophique révèle une autophagie dans les adipocytes matures. Autophagie. 2010 Août; 6 (6): 754-63.
- Liu L, Brown D, McKee M, et al. La délétion de la cavine / PTRF entraîne une perte globale des cavéoles, une dyslipidémie et une intolérance au glucose. Métabolisme cellulaire. 2008 Oct; 8(4):310-7.
- Garg A, Wilson R, Barnes R, et al. Un gène de la lipodystrophie généralisée congénitale correspond au chromosome 9q34 humain. Le Journal d’endocrinologie clinique et du métabolisme. 1999 Sep; 84(9):3390-4.
- Vogel P, lire R, Hansen G, et al. Pathologie de la lipodystrophie généralisée congénitale chez les souris Agpat2-/-. Pathologie vétérinaire. 2011 Mai; 48 (3): 642-54.
- Cortes VA, Curtis DE, Sukumaran S, et al. Molecular mechanisms of hepatic stéatosis and insulin resistance in the AGPAT2-deficient mouse model of congénital generalized lipodystrophy. Métabolisme cellulaire. 2009 Fév; 9 (2): 165-76.
- Kim CA, Delepine M, Boutet E, et al. Association d’une mutation homozygote non-sens de la cavéoline-1 avec une lipodystrophie congénitale Berardinelli-Seip. J Clin Endocrinol Metab. 2008 Avril; 93(4): 1129-34.
- Rajab A, Straub V, McCann LJ, et al. Arythmie cardiaque fatale et syndrome du QT long dans une nouvelle forme de lipodystrophie généralisée congénitale avec ondulation musculaire (CGL4) due à des mutations PTRF-CAVINE. Génétique PLoS. 12 Mars 2010; 6 (3): e1000874.
- Payne F, Lim K, Girousse A, et al. Mutations perturbant la voie de la phosphatidylcholine de Kennedy chez les humains atteints de lipodystrophie congénitale et de stéatose hépatique. Proc Natl Acad Sci U S A. 2014 17 juin; 111 (24): 8901-6.
- Dyment DA, Gibson WT, Huang L, et al. Les mutations bialléliques à PPARG provoquent une lipodystrophie congénitale généralisée similaire au syndrome de Berardinelli-Seip. Eur J Med Genet. 2014 Sept; 57 (9): 524-6.
- Garg A. Revue clinique #: Lipodystrophies: troubles génétiques et acquis de la graisse corporelle. Le Journal d’endocrinologie clinique et du métabolisme. 2011 Nov; 96 (11): 3313-25.
- Lima JG, Nobrega LH, de Lima NN, et al. Données cliniques et de laboratoire d’une grande série de patients atteints de lipodystrophie généralisée congénitale. Diabétol Metab Syndr. 2016; 8: 23.
- Lima GJ, Lima NN, Oliveira CF, et al. Hernie ombilicale chez les patients atteints de syndrome de Berardinelliseip: Est-ce vraiment une Hernie. J Clin Mol Endocrinol. 2015; 1(1): 3.
- Brown RJ, Araujo-Vilar D, Cheung PT, et al. Le Diagnostic et la Prise en Charge des Syndromes de Lipodystrophie: Un Guide de pratique Multisociété. J Clin Endocrinol Metab. 2016 Déc; 101 (12): 4500-11.
- Lima JG, Nobrega LH, Lima NN, et al. La Densité Osseuse chez les Patients Atteints De Lipodystrophie Congénitale Berardinelli-Seip Est Plus Élevée dans les Sites Trabéculaires et chez les Patients de Type 2. J Clin Densitom. 25 novembre 2016.
- Simha V, Garg A. Phenotypic heterogeneity in body fat distribution in patients with congénital generalized lipodystrophy caused by mutations in the AGPAT2 or seipin genes. J Clin Endocrinol Metab. 2003 Nov; 88 (11): 5433-7.
- Musso C, Cochran E, Javor E, et al. L’effet à long terme de la leptine humaine méthionyl recombinante sur l’hyperandrogénie et la fonction menstruelle chez la femme et la fonction hypophysaire chez les patients lipodystrophiques hypoleptinémiques masculins et féminins. Métabolisme. 2005 Fév; 54(2): 255-63.
- Jiang M, Gao M, Wu C, et al. L’absence de seipine testiculaire provoque un syndrome de tératozoospermie chez les hommes. Proc Natl Acad Sci U S A. 2014 13 mai; 111 (19): 7054-9.
- Mitchell O, Feldman DM, Diakow M, et al. La physiopathologie de la thrombocytopénie dans les maladies chroniques du foie. Hepat Med. 2016; 8: 39-50.
- Joubert M, Jagu B, Montaigne D, et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes. 2017 Apr; 66(4): 1030-40.
- Lima JG, Lima NN, Lima RLM, et al. Glargine U300 Insulin as a Better Option than Degludec U100 to Treat a Congenital Generalized Lipodystrophy Patient. Clin Diabetes Res. 2017; 1(1).