ConSurf
Introduction
ConSurf est un outil bioinformatique permettant d’estimer la conservation évolutive des positions des acides aminés/nucléiques dans une molécule protéine/ADN/ARN sur la base des relations phylogénétiques entre séquences homologues. Le degré de conservation évolutive d’une position d’acide aminé (ou nucléique) dépend fortement de son importance structurelle et fonctionnelle; les positions évoluant rapidement sont variables tandis que les positions évoluant lentement sont conservées. Ainsi, l’analyse de conservation des positions parmi les membres d’une même famille peut souvent révéler l’importance de chaque position pour la structure de la protéine (ou de l’acide nucléique) ou function.In En conséquence, le taux d’évolution est estimé sur la base de la relation évolutive entre la protéine (ADN / ARN) et ses homologues et compte tenu de la similitude entre les acides aminés (nucléiques) tels que reflétés dans la matrice de substitutions. L’un des avantages de ConSurf par rapport aux autres méthodes est le calcul précis du taux d’évolution en utilisant soit une méthode bayésienne empirique, soit une méthode du maximum de vraisemblance (ML).
Méthodologie
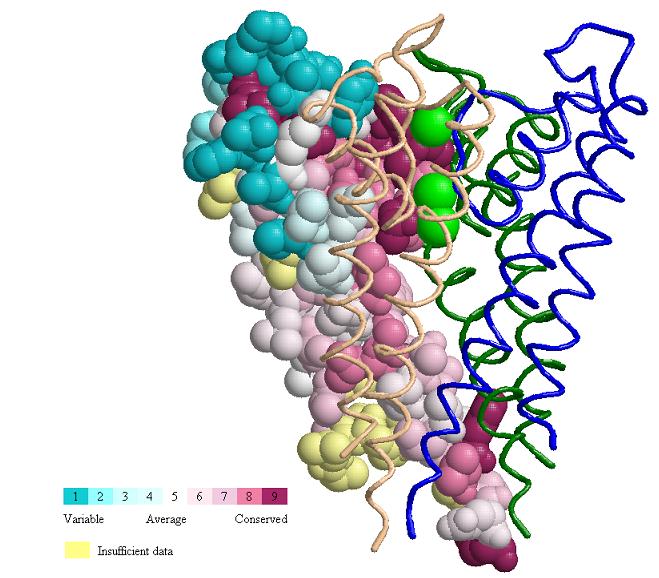
Compte tenu de la séquence d’acides aminés ou nucléiques (pouvant être extraite de la structure 3D), ConSurf effectue une recherche de séquences homologues proches en utilisant BLAST (ou PSI-BLAST). L’utilisateur peut sélectionner l’une de plusieurs bases de données et spécifier des critères pour définir des homologues. L’utilisateur peut également sélectionner les séquences souhaitées à partir des résultats de BLAST. Les séquences sont regroupées et des séquences très similaires sont supprimées à l’aide de CD-HIT. Un alignement de séquences multiples (MSA) des séquences homologues est construit à l’aide de MAFFT, PRANK, T-COFFEE, MUSCLE (par défaut) ou CLUSTALW. Le MSA est ensuite utilisé pour construire un arbre phylogénétique à l’aide de l’algorithme de jointure de voisins implémenté dans le programme Rate4Site. Les scores de conservation spécifiques à la position sont calculés à l’aide des algorithmes bayésiens empiriques ou ML. Les scores de conservation continue sont divisés en une échelle discrète de neuf grades pour la visualisation, des positions les plus variables (grade 1) turquoise de couleur, aux positions moyennement conservées (grade 5) blanc de couleur, aux positions les plus conservées (grade 9) marron de couleur. Les scores de conservation sont projetés sur la séquence protéine/nucléotide et sur le MSA
 |
 |