Frontiers in Neurology
Introduction
La neuroacanthocytose est un terme supérieur pour un groupe de syndromes rares caractérisés par des symptômes neurologiques associés à des globules rouges déformés hérissés (acanthocytes). Ce rapport se concentre sur la chorée-acanthocytose (ChAc), qui est une maladie orpheline avec environ 1 000 individus affectés dans le monde, causée par des mutations autosomiques récessives du gène VPS13A (vacuolar protein sorting 13 homologa) sur le chromosome 9q (1). La maladie évolue progressivement, les causes de mortalité accrue peuvent être des fonctions motrices diminuées en corrélation avec des conditions de risque telles que la dysphagie, mais des décès soudains et inattendus ont également été décrits (2, 3). Jusqu’à présent, il n’y a pas de traitement causal.
La coexistence suspecte d’acanthocytose et de troubles du mouvement a été signalée pour la première fois dans les années 1970 par Levine et Critchley (4, 5) et a depuis été acceptée comme la caractéristique principale de la maladie. Cependant, avec notre rapport de cas, nous décrivons un phénotype clinique rare de ChAc dépourvu de troubles du mouvement évidents, suggérant une plus grande variété de présentations cliniques. Les outils de diagnostic doivent relever ces défis pour établir le bon diagnostic, nous proposons ici la neuroimagerie moléculaire comme méthode clé.
Rapport de cas
Le patient index masculin a présenté pour la première fois à l’âge de 25 ans deux crises cloniques toniques bilatérales non provoquées. Il n’y avait pas d’antécédents médicaux. L’IRM cérébrale et l’EEG à cet âge étaient normaux et aucun traitement n’a été initié en raison de la réticence du patient et d’événements peu fréquents. Cependant, le patient avait des crises en cours se présentant maintenant comme une épilepsie partielle. Il a décrit des sensations récurrentes de vertiges soudains qui ont été diagnostiquées comme une aura vertigineuse. De plus, il a eu des crises dyscognitives dans lesquelles il a montré soit (a) une insensibilité et une récitation de prières turques, (b) des automatismes oraux, soit (c) un tripotage des mains et des sons — le tout évoluant en partie en crises cloniques toniques. Vidéo – L’EEG a maintenant montré des complexes locaux d’ondes de pic à la fois dans le lobe temporal droit et gauche. Conformément à la sémiologie de la crise, le diagnostic d’épilepsie bilatérale du lobe temporal a été posé et des médicaments avec du lévétiracétam ont été lancés.
À l’âge de 31 ans, les crises n’étaient pas entièrement supprimées, dans le cadre d’autres diagnostics de l’épilepsie du lobe temporal, le patient a subi un TEP FDG (Figure 1) qui a montré un hypométabolisme mésiotemporal bilatéral, conforme à l’origine de la crise mésiotemporale. Cependant, il y avait aussi un hypométabolisme striatal marqué et très inhabituel qui a soulevé la suspicion d’un trouble du mouvement neurodégénératif. Par conséquent, une évaluation de suivi approfondie a été lancée (voir figure 2). Une FP-CIT-SPECT a été réalisée qui a donné une perte modérément bilatérale de la disponibilité du transporteur striatal de dopamine, en ligne avec une baisse de l’intégrité nigrostriatale (Figure 1). Une IRM à haute résolution présente maintenant une atrophie bilatérale du noyau caudatus (Figure 3). L’examen neurologique a montré un schéma de démarche lié discret et une hypomimie, mais aucune autre affection du système moteur extrapyramidal et aucun symptôme bulbaire comme la dystonie alimentaire. Mis à part un état réflexe bas sur les membres inférieurs, pour lequel des études de conduction nerveuse ont confirmé une polyneuropathie axonale sensorimotrice, l’examen neurologique était normal. Les symptômes neuropsychologiques comprenaient des aspects d’un trouble anxieux et le patient a signalé des difficultés avec une perte de mémoire à court terme. Les tests neuropsychologiques n’ont cependant pas confirmé un dysfonctionnement mnésique manifeste, mais plutôt une distraction non spécifique, qui aurait pu en outre être renforcée par une suppression insuffisante des crises à l’époque. Les tests de laboratoire étaient négatifs pour toute épilepsie symptomatique (p. ex., anticorps d’encéphalite auto-immune, anticorps antineuronaux). Le liquide céphalo-rachidien n’a montré aucun signe d’inflammation mais une augmentation modérée des protéines (765 mg / l). L’analyse des amines biogènes dans le liquide céphalo-rachidien a révélé une élévation de la glutamine que nous avons associée à des crises récentes. Dans le sang périphérique, 0,4% des acanthocytes ont été trouvés. Les tests sérologiques pour la maladie de Wilson étaient négatifs. Une élévation persistante de la créatine kinase (entre 1 000 et 3 000 U / l) a été remarquée, ce qui a conduit au diagnostic présumé d’un trouble mitochondrial. Pour des investigations plus approfondies, un test de lactate ischémique a été effectué qui a montré des résultats normaux. Une biopsie musculaire du M. gastrocnemicus a été réalisée, mais l’analyse de la fonction mitochondriale et de la chaîne respiratoire était normale.
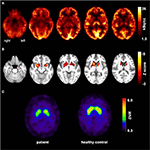
Figure 1. Résultats d’études d’imagerie moléculaire. Les images transaxiales FDG-TEP (A) ont montré non seulement un léger hypométabolisme mésiotemporal bilatéral (conforme à l’origine de la crise mésiotemporale), mais également un hypométabolisme striatal marqué qui s’est avéré très significatif par rapport aux témoins sains. Un examen FP-CIT-SPECT supplémentaire (C) a également révélé une perte modérée de transporteurs de dopamine striatale, témoin d’un déclin de l’intégrité nigrostriatale (un contrôle sain adapté à l’âge est montré à des fins de comparaison; DVR, rapport de volume de distribution).

Figure 2. Évolution clinique et diagnostique du patient index.
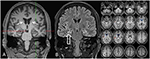
Figure 3. L’IRM montre une atrophie bilatérale discrète de la tête caudée et un hippocampe droit plus petit et hyperintensif indiquant une sclérose de l’hippocampe droit. L’hippocampe gauche est quelque peu hyperintense mais pas atrophique. L’atrophie bilatérale de la tête caudée est confirmée par une analyse CVR, dans laquelle les couleurs bleues montrent une diminution du volume de liquide céphalo-rachidien, respectivement (C).
Les antécédents familiaux ont révélé une consanguinité de ses parents (cousins germains), qui eux-mêmes n’avaient pas d’antécédents médicaux significatifs. Parmi ses six frères et sœurs, deux (âgés de 20 à 30 ans) souffraient également de crises générales (voir Figure 4). Les dossiers médicaux de ses frères et sœurs n’étaient pas à notre disposition.
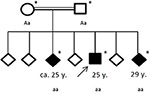
Figure 4. Pedigree de la famille touchée, avec l’âge de la première crise d’épilepsie en années (y). Flèche : brevet d’index. Astérisque : test génétique effectué. Aa, hétérozygote; aa, homozygote pour environ 4326 T > A (p. Tyr1442*).
Dans l’hypothèse d’un trouble du mouvement neurodégénératif héréditaire, le patient et sa famille ont été référés pour des tests génétiques. Le séquençage des exomes a révélé une nouvelle mutation tronquée homozygote c. 4326 T > A (p. Tyr1442*) dans le gène ChAc VPS13A chez les trois frères et sœurs affectés. Les parents consanguins ont tous deux été détectés hétérozygotes. Aucune autre variante ou mutation n’a été identifiée dans le séquençage de l’exome.
En effet, lors d’un suivi à l’âge de 33 ans, le patient index n’avait encore que des symptômes extrapyramidaux marginaux et aucun n’était présent chez aucune de ses sœurs. Il donnait maintenant l’impression d’une légère rigidité seulement sous l’amorçage du membre supérieur droit, et d’une légère bradykinésie des membres supérieurs. Sous traitement par le lévétiracétam et le lacosamide, le patient n’a pas eu de convulsions.
Discussion
Dans le cas présent, nous démontrons que ChAc peut se manifester cliniquement avec une épilepsie du lobe temporal dépourvue de symptômes moteurs significatifs en raison d’une nouvelle mutation correspondante dans le gène VPS13A.
L’évolution de la maladie est généralement caractérisée par un trouble du mouvement progressif (y compris la chorée, la dystonie, le parkinsonisme), des modifications cognitives et psychiatriques et des symptômes myopathiques avec des marqueurs sérologiques d’acanthocytose et d’hyperCKémie. Des convulsions ont déjà été notées comme symptôme de ChAc (6) mais les troubles du mouvement sont toujours considérés comme le symptôme clé. Il est prouvé que l’épilepsie, et plus précisément l’épilepsie provenant du lobe temporal, pourrait être un phénotype sous-estimé de ChAc. Peluso et coll. a rapporté un patient similaire au nôtre, qui, même à l’âge de 46 ans, ne présentait pas de troubles du mouvement tout en présentant des résultats de neuroimagerie indiquant une atteinte des ganglions de la base (7). Conformément à cela, Scheid et al. a décrit trois patients souffrant d’épilepsie et de sclérose du lobe temporal mésial (sur la base d’études IRM) comme symptôme prédominant et ont reçu un diagnostic de ChAc (8). De plus, Mente et coll. résultats d’autopsie récemment fournis d’un patient de la ChAc présentant non seulement une atrophie des ganglions de la base, mais également une sclérose de l’hippocampe (9). Dans la lignée de notre cas, l’implication bilatérale de l’hippocampe semble être une constatation courante (10, 11). La relation exacte entre les crises et la sclérose est cependant toujours une question de poule ou d’œuf. Le rôle corrélatif de la choréine dans le développement structurel de l’hippocampe n’est pas encore entièrement compris. Il est intéressant de noter que dans un modèle murin, un changement structurel de la ChAc a été observé, car l’expression de la protéine hippocampique du récepteur GABA (A) gamma 2 et de sa protéine d’ancrage choréine ont été augmentées (12).
En routine clinique, ce phénotype peut encore être difficile pour trouver le bon diagnostic. Le premier indice crucial d’une maladie neurodégénérative chez notre patient était une baisse importante du métabolisme du glucose striatal et une baisse modérée de l’intégrité nigrostriatale sur FDG-PET et FP-CIT-SPECT, respectivement. La petite collection de résultats d’imagerie fonctionnelle, basée sur la description de la série de cas, montre que des altérations du métabolisme et un dysfonctionnement dopaminergique se produisent principalement dans le noyau caudé et le putamen (13). L’IRM cérébrale a révélé une atrophie du noyau caudatus au cours de la maladie, ce qui a été décrit comme une découverte typique chez ChAc (14). En conséquence, les caractéristiques neurologiques distinctes comprennent généralement la chorée, la dystonie nourricière, les dyskinésies orofaciolingues et les tics, ainsi que d’autres symptômes d’affection des ganglions de la base tels que le parkinsonisme et la dystonie (3). Il est tentant de spéculer que les changements striataux observés chez notre patient étaient toujours en dessous du seuil de provoquer des symptômes (c.-à-d. des résultats d’imagerie prodromique), qui peuvent finalement survenir à mesure que la maladie progresse. Nous encourageons donc l’imagerie structurale et moléculaire comme outil de diagnostic sensible dans cette maladie orpheline.
Dans ChAc, il existe des descriptions de dénombrements d’acanthocytes entre 5 et 50% (3), mais il existe quelques rapports de cas qui démontrent que les acanthocytes peuvent apparaître seulement tard dans l’évolution de la maladie (15), peuvent être très faibles, voire totalement absents (16). Par conséquent, les acanthocytes ne doivent pas être considérés comme obligatoires pour poser le diagnostic. De plus, l’épilepsie peut retarder le diagnostic car elle dissimule d’autres symptômes typiques de la ChAc. En particulier, les symptômes psychiatriques tels que les changements de personnalité et la détérioration cognitive, qui sont très fréquents, peuvent être mal interprétés et attribués à des effets secondaires liés au médicament (c.-à-d. du lévétiracétam) ou à des crises convulsives. Une hypercémie secondaire est observée après des crises cloniques toniques générales et une élévation persistante peut être négligée.
Notre cas met également en évidence l’avantage crucial du séquençage des exomes pour établir un diagnostic correct, en particulier dans les maladies rares ou lorsque les symptômes sont vagues. La famille des gènes VPS13 (A–D) des mammifères présente un intérêt croissant et des mutations ont été identifiées dans d’autres maladies neurologiques telles que la maladie de Parkinson autosomique récessive ou l’ataxie spinocérébelleuse autosomique récessive (17). Les mutations récessives de VPS13D provoquent des troubles du mouvement chez l’enfant (18). La physiopathologie sous-jacente de ChAc n’est pas encore complètement déchiffrée, mais il a été démontré que VPS13A code la protéine choréine qui est exprimée de manière ubiquitaire dans le cerveau et dans un grand nombre d’autres tissus (19). Des études révèlent qu’il participe aux voies de signalisation qui régulent l’architecture cytosquelettique, l’exocytose et la survie cellulaire (20). Une autre mutation qui a été décrite comme présentant un phénotype dominé par les crises chez 9 patients atteints de ChAc est la mutation c.2343del du gène VPS13A (21). Il est raisonnable de supposer que des mutations spécifiques dans le même gène entraînent une certaine aberration de la choréine qui provoque un phénotype unique, par exemple en affectant différentes zones du cerveau. Cependant, la physiopathologie sous-jacente n’est encore que spéculative et une documentation plus approfondie des mutations causales sera intéressante. Nous montrons ici que la mutation tronquée c. 4326 T > A (p. Tyr1442 *), qui n’a pas été décrite auparavant, aboutit à un phénotype similaire avec une épilepsie prédominante chez tous les membres de la famille affectés.
Remarques finales
L’épilepsie (en particulier l’épilepsie bilatérale du lobe temporal) est susceptible d’être un phénotype prédominant de la ChAc. Par conséquent, une recherche de drapeaux rouges supplémentaires (tels que l’hypercémie, les antécédents familiaux, la polyneuropathie) doit être soigneusement effectuée. Dans les cas suspects, la recherche d’acanthocytes dans le sang et l’IRM cérébrale doit être ajoutée car ces outils sont largement disponibles. En cas de doute, le diagnostic doit être complété par FDG-PET et / ou FP-CIT-SPECT car ils sont sensibles à la détection de l’atteinte striatale. Dans les épilepsies autosomiques récessives avec des résultats indicatifs pour une maladie neurodégénérative, un test de gène unique VPS13A ou un Western blot de choréine doit ensuite être utilisé pour établir le bon diagnostic. Ce dernier devrait également être pris en compte dans d’autres troubles neurodégénératifs sans étiologie génétiquement définie.
Déclaration d’éthique
Le consentement éclairé écrit du patient a été obtenu pour participer à l’étude. Le consentement éclairé écrit du patient a été obtenu pour la publication de ce rapport de cas.
Contributions des auteurs
JW, MR et SK ont participé à la prise en charge des patients. JW a écrit la première ébauche du manuscrit. LF, HU et PM ont préparé des chiffres. Tous les auteurs ont contribué à la révision du manuscrit, ont lu et approuvé la version soumise.
Déclaration de conflit d’intérêts
HU est actionnaire de Veobrain Gmbh, une spin-off du Centre Médical Universitaire de Fribourg.
Les autres auteurs déclarent que la recherche a été menée en l’absence de relations commerciales ou financières pouvant être interprétées comme un conflit d’intérêts potentiel.
1. Il s’agit de l’une des principales sources de pollution de l’air dans le monde. La neuropsychiatrie des syndromes de neuroacanthocytose. Neurosci Biobehav Rev. (2011) 35:1275-83. doi: 10.1016/ j.neubiorev.2011.01.001
Résumé PubMed / CrossRef Texte intégral / Google Scholar
2. Walker RH. Gestion des syndromes de neuroacanthocytose. Tremblement Autre Hyperkinet. Mov. (2015) 5:346. doi: 10.7916 /D8W66K48
Résumé publié / Texte intégral croisé / Google Scholar
3. Velayos Baeza A, Dobson-Stone C, Rampoldi L, Bader B, Walker RH, Danek A, et al. Chorée-Acanthocytose. GeneReviews®. Seattle, WA: Université de Washington (1993).
4. Critchley EM, Clark DB, Wikler A. Acanthocytose et Trouble neurologique sans bétalipoprotéinémie. Arch Neurol. (1968) 18:134–40.
Résumé PubMed
5. Levine IM, Estes JW, Looney JM. Maladie neurologique héréditaire avec acanthocytose. Un nouveau syndrome. Arch Neurol. (1968) 19:403–9.
Résumé PubMed / Google Scholar
6. Hardie RJ, Pullon HW, Harding AE, Owen JS, Pires M, Daniels GL, et al. Neuroacanthocytose. une étude clinique, hématologique et pathologique de 19 cas. Brain (1991) 114 (Pt 1A): 13-49.
Résumé PubMed / Google Scholar
7. Peluso S, Bilo L, Esposito M, Antenora A, Rosa ADR, Pappata S, et al. Chorée-acanthocytose sans chorée: expansion du phénotype clinique. Maladie de Parkinson. (2017) 41:124–6. doi: 10.2016 / j. parkreldis.2017.05.013
Résumé PubMed / Texte intégral CrossRef / Google Scholar
8. Scheid R, Bader B, Ott DV, Merkenschlager A, Danek A. Développement de l’épilepsie du lobe temporal mésial dans la chorée-acanthocytose. Neurology (2009) 73:1419-22. doi: 10.1212/WNL.0b013e3181bd80d4
Résumé publié / Texte intégral croisé / Google Scholar
9. Mente K, Kim SA, Grunseich C, Hefti MM, Crary JF, Danek A, et al. Sclérose de l’hippocampe et épilepsie du lobe temporal mésial dans la chorée-acanthocytose: un cas avec évaluation clinique, pathologique et génétique. Neuropathol Appl Neurobiol. (2017) 43:542–6. doi: 10.1111/nan.12403
Résumé PubMed / Texte intégral CrossRef / Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert A, la Fougère C, Noachtar S, et al. Épilepsie bilatérale du lobe temporal confirmée par EEG intracrânien dans la chorée-acanthocytose. Saisie (2011) 20:340-2. doi: 10.1016 /j. saisie.2010.12.007
Résumé PubMed / Texte intégral CrossRef / Google Scholar
11. Marson AM, Bucciantini E, Gentile E, Geda C. Neuroacanthocytose: résultats cliniques, radiologiques et neurophysiologiques dans une famille italienne. Neurol Sci. (2003) 24:188–9. doi: 10.1007/s10072-003-0123-1
Résumé PubMed / Texte intégral CrossRef / Google Scholar
12. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, et al. Une carence en choréine entraîne une régulation à la hausse du récepteur de la géphyrine et du GABA (A). Biochem Biophys Res Commun. (2006) 351:438–42. doi: 10.1016/j.bbrc.2006.10.070
Résumé PubMed / Texte intégral CrossRef / Google Scholar
13. Ehrlich DJ, Walker RH. Neuroimagerie fonctionnelle et chorée: une revue systématique. J Clin Mov Disord. (2017) 4:8. doi: 10.1186/s40734-017-0056-0
Résumé PubMed / Texte intégral CrossRef / Google Scholar
14. Connolly BS, Hazrati L-N, Lang AE. Neuropathological Findings in chorea-acanthocytosis: new insights into mechanisms underlying Parkinsonism and seizures. Acta Neuropathol. (2014) 127:613–5. doi: 10.1007/s00401-013-1241-3
Résumé PubMed / Texte intégral CrossRef / Google Scholar
15. Sorrentino G, De Renzo A, Miniello S, Nori O, Bonavita V. Apparition tardive des acanthocytes au cours de la chorée-acanthocytose. J Neurol Sci. (1999) 163:175–8.
Résumé PubMed / Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Choréo-acanthocytose sans acanthocytes. Revue Neurol. (2010) 166:100–3. doi: 10.1016/ j.neurol.2009.03.005
CrossRef Texte intégral / Google Scholar
17. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. La perte de la fonction VPS13C dans le parkinsonisme autosomique récessif provoque un dysfonctionnement mitochondrial et augmente la mitophagie dépendante de PINK1/Parkin. Je suis J Hum Genet. (2016) 98:500–13. doi: 10.1016/j.ajhg.2016.01.014
Résumé PubMed / Texte intégral CrossRef / Google Scholar
18. Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M, et al. Les mutations récessives de > VPS13D provoquent des troubles du mouvement chez l’enfant. Ann Neurol. (2018) 83:I6. doi: 10.1002/ ana.25204
Résumé PubMed / Texte intégral CrossRef / Google Scholar
19. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, et al. Distribution in vivo et localisation de la choréine. Biochem Biophys Res Commun. (2007) 353:431–5. doi: 10.1016/j.bbrc.2006.12.059
Résumé PubMed / Texte intégral CrossRef / Google Scholar
20. Lang F, Pelzl L, Schöls L, Hermann A, Föller M, Schäffer TE, et al. Neurones, érythrocytes et au-delà – les diverses fonctions de la choréine. Neurosignaux (2017) 25:117-26. doi: 10.1159/000485457
Résumé PubMed / Texte intégral CrossRef / Google Scholar
21. Benninger F, Afawi Z, Korczyn AD, Oliver KL, Pendziwiat M, Nakamura M, et al. Convulsions en tant que symptôme présent et important dans la chorée-acanthocytose avec mutation du gène VPS13A c.2343del. Épilepsie (2016) 57:549-56. doi: 10.1111/epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar