Frontiers in Pediatrics
- Introduction
- Syndrome du QT long
- Les mutations LQTS1
- LQTS2
- LQTS3
- LQTS4
- LQTS5
- LQTS6
- LQTS7
- LQTS8
- LQTS9 et LQTS10
- LQTS11
- LQTS12
- LQTS13
- LQTS acquis
- Caractéristiques électrocardiographiques dans les Trois formes courantes du syndrome du QT long
- Génotype-Phénotype
- La prise en charge clinique des LQTS
- LQTS: Perspective saoudienne
- Déclaration de conflit d’intérêts
- Remerciements
Introduction
Les arythmies cardiaques familiales ou héréditaires comprennent des pourcentages significatifs d’arythmies et sont également causales à la mort cardiaque subite (DCS) (1, 2). Au cours des deux dernières décennies, les scientifiques et les cliniciens ont fourni d’énormes efforts pour démêler les mécanismes complexes et complexes des arythmies familiales congénitales (1-8). Pour comprendre le mécanisme de l’arythmogenèse, nous devons connaître les bases de la structure cellulaire cardiaque et leurs propriétés électrophysiologiques. Les myocytes cardiaques sont les principales cellules fonctionnelles du cœur et ils sont largement couplés de sorte que les impulsions se propagent rapidement et uniformément. Les cardiomyocytes sont séparés les uns des autres par une limite spécialisée appelée disque intercalé; les protéines de jonction d’espace, les desmosomes cardiaques et les canaux ioniques sont situés dans le disque intercalé (9). Les jonctions lacunaires sont constituées de connexines serrées qui permettent l’échange intercellulaire de petites molécules et permettent également de faire circuler les courants excitateurs d’une cellule vers sa cellule voisine. Les desmosomes ainsi que les jonctions adhérentes sont responsables des attachements mécaniques des cardiomyocytes individuels. Tous ces composants dans les disques intercalés sont séparés séquentiellement et chaque composant exerce sa fonction unique; la perturbation de l’un des composants affecte la fonction des autres composants, ce qui prédispose le cœur à développer des arythmies (9-11). Les canaux ioniques cardiaques sont des complexes protéiques formant des pores qui fournissent un mouvement intérieur et extérieur coordonné et complexe des courants ioniques à travers les membranes cellulaires, essentiel à la génération et à la propagation du rythme cardiaque. Le syndrome du QT long (LQTS), le syndrome du QT court (SQTS), le syndrome des sinus malades (SSS), le défaut de conduction cardiaque (CCD), le SRB, la tachycardie ventriculaire polymorphe catécholaminergique (CPVT), le syndrome de repolarisation précoce (ERS) et la fibrillation auriculaire familiale (FA) sont les channelopathies cardiaques actuellement connues, qui pourraient survenir en raison d’un défaut unique ou multiple dans les gènes liés à la génération et à la propagation du rythme cardiaque.
Dans cette revue, nous décrirons exclusivement sur les arythmies liées au LQTS, sa physiopathologie et sa prise en charge clinique actuellement disponible. Nous avons été pionniers dans l’élucidation de la pathologie génétique dans les arythmies cardiaques familiales en Arabie saoudite (1, 12-14), nous menons toujours des enquêtes cardiogénétiques en Arabie saoudite, à la fin de cette revue, nous discuterons des connaissances actuellement disponibles sur les résultats génétiques et cliniques obtenus auprès de plusieurs familles saoudiennes ayant des antécédents de syncope et de SCD. Nous ajouterons également les données récemment publiées sur les TQL d’une équipe de l’Hôpital spécialisé et du Centre de recherche King Faisal, à Riyad (15).
Syndrome du QT long
Le LQTS congénital est un trouble héréditaire défini par l’allongement de l’intervalle QT sur électrocardiogramme (ECG). Les patients présentant toutes les formes de LQTS sont prédisposés à la tachyarythmie ventriculaire, aux torsades de pointes (TdP) conduisant à une syncope récurrente, ou SCD. Dans de nombreux cas, la syncope ou la mort subite pourrait être la première et la seule manifestation. Le TQL affecte environ 1 personne sur 2 000 dans le monde (16). La caractéristique du LQTS est l’allongement de l’intervalle QT sur l’ECG (corrigé pour la fréquence cardiaque, c’est-à-dire QTc). Les valeurs normales de QTc sont de 440 ms chez les hommes et de 450 ms chez les femmes. Chez les enfants, les valeurs dépendantes de l’âge et du sexe sont pertinentes. Les recommandations consensuelles récentes pour le diagnostic de LQTS sont les suivantes (17, 18):
1. LQTS est diagnostiqué:
a. En présence d’un score de risque LQTS ≥3,5 en l’absence de cause secondaire d’allongement de l’intervalle QT et / ou
b. En présence d’une mutation pathogène sans équivoque dans l’un des gènes LQTS ou
c. En présence d’un intervalle QT corrigé de la fréquence cardiaque à l’aide de la formule de Bazett (QTc) ≥500 ms dans un électrocardiogramme répété à 12 dérivations et en l’absence de cause secondaire d’allongement de l’intervalle QT.
2. Les LQTS peuvent être diagnostiqués en présence d’un QTc compris entre 480 et 499 ms dans des ECG à 12 dérivations répétées chez un patient présentant une syncope inexpliquée en l’absence de cause secondaire d’allongement de l’intervalle QT et en l’absence de mutation pathogène.
Les patients avec une durée QTc ≥500 ms avaient une probabilité cumulative significativement plus élevée de subir leur première syncope par rapport aux patients avec une durée QTc < 500 ms de la naissance à l’âge de 20 ans (19). Mais, chez les patients ayant connu 2, 3 et 4 épisodes de syncope, le risque d’un épisode de syncope ultérieur était pratiquement identique entre les patients ayant une durée QTc étroite ou prolongée (19). La base moléculaire des LQTS est hétérogène et à ce jour, des mutations dans 13 gènes différents ont été décrites causales aux LQTS (1-8, 19, 20). Un défaut génétique est généralement trouvé chez 70% des patients LQTS dans l’un de ces 13 gènes (1-8, 19, 20). Parmi les 13 types différents de LQTS actuellement connus, les plus courants sont les LQTS1, LQTS2 et LQTS3, en raison de défauts dans les gènes du canal ionique cardiaque, KCNQ1, KCNH2 et SCN5A, respectivement. Quatre-vingt-dix pour cent de toutes les mutations causales du LQTS se trouvent dans ces trois gènes (1-8, 19, 20). Les mutations dans les 10 gènes restants sont rares et ne représentent que110% de toutes les mutations LQTS actuellement connues.
Le syndrome du QT long est généralement une maladie autosomique dominante, mais, occasionnellement, des mutations multiples dans un seul gène ou dans des gènes différents peuvent être trouvées chez 5 à 10% des patients atteints de LQTS (21, 22). Les patients présentant plusieurs mutations pourraient présenter un QTc plus long que ceux présentant une seule mutation et ces patients présentent également un risque accru de 3,5 fois plus élevé d’événements cardiaques mettant leur vie en danger (21, 22)
Les mutations LQTS1
du gène KCNQ1 sont la forme la plus courante de tous les LQTS et sont appelées LQTS de type 1 (LQTS1). Cinquante pour cent de toutes les mutations causales du LQTS se trouvent dans le gène KCNQ1 (20). KvLQT1 (également appelée Kv7.1) est une protéine fabriquée par le gène KCNQ1 et la protéine forme des tétramères dans le réticulum endoplasmique à l’intérieur des cellules, qui se co-assemble putativement avec la protéine minK (codée par KCNE1) et sont ensuite transportées vers la membrane plasmique des myocytes cardiaques, où elles médient un courant d’activation lente qui accélère la repolarisation du potentiel d’action dans les tissus cardiaques, ce courant est connu sous le nom d’IKs (Figure 1). Les mutations causales KCNQ1 de LQTS1 sont pour la plupart des mutations faux sens et, dans de rares cas, il pourrait s’agir de mutations à décalage de cadre dans la région C-terminale (23). L’arythmie cardiaque chez les porteurs de la mutation KCNQ1 est déclenchée par des pulsions adrénergiques, par exemple le stress émotionnel, l’effort physique, la plongée, la natation (24-26). Les patients présentant des mutations dans les domaines transmembranaires de KvLQT1 présentent un risque plus élevé d’événements cardiaques liés au LQTS et présentent une plus grande sensibilité à la stimulation sympathique (26, 27). Les patients présentant des mutations faux sens présentent un risque accru par rapport aux patients présentant des mutations non sensorielles ou tronquées (27, 28). La variation du 3′-UTR dans le gène KCNQ1 affecte également de manière significative la sensibilité à l’arythmie, probablement en ayant un impact sur l’expression du gène (29). Les patients présentant des arythmies dues à des mutations KCNQ1 répondent assez bien aux β-bloquants, mais certains patients pourraient toujours être moins réactifs ou même résistants à ce médicament. Dans un article récent, Barsheshet et al. (30) ont affirmé que les patients présentant des mutations en dehors de la région de la boucle cytoplasmique (boucle c) dans le KvLQT1 sont moins sensibles aux β-bloquants.
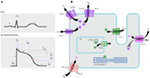
Figure 1. Courants ioniques contribuant au potentiel d’action ventriculaire (A) et représentation schématique d’un cardiomyocyte affichant (uniquement) les protéines impliquées dans la pathogenèse des syndromes d’arythmie héréditaire (B). En (A), le potentiel d’action est aligné avec son temps d’action approximatif pendant l’ECG. En (B), l’ankyrine-B, une protéine adaptatrice impliquée dans le syndrome du QT long de type 4, n’est pas représentée.
Les mutations homozygotes ou hétérozygotes composées du gène KCNQ1, qui pourraient provoquer la forme récessive de la maladie, le syndrome de Jervell et de Lange-Nielsen (JLNS), de type 1 (31), sont assez rares. Les patients atteints de JLNS souffrent également d’arythmies cardiaques sévères et de surdité (31, 32). Les patients présentant des mutations KCNQ1 causales aux JLN ne présentent généralement pas d’IKs fonctionnels (28, 31-33). Il est possible que certains patients n’aient pas de surdité malgré des mutations homozygotes ou hétérozygotes composées dans KCNQ1, ces cas sont appelés LQTS1 autosomique récessif (13, 32). Chez ces patients, une petite quantité de courant IKs fonctionnel (< 10% de l’IKs total) pourrait encore être présente, ce qui maintient la fonction auditive, mais leurs anomalies du rythme cardiaque sont tout aussi graves que chez les patients JLNS (13, 32).
LQTS2
Ce type de LQTS est également répandu que le LQTS1, représentant 35 à 40% des patients atteints de LQTS avec une mutation détectable (1, 20, 24, 25). KCNH2 code pour la protéine HERG (Kv11.1), qui est la sous-unité α du courant K+ du redresseur retardé à activation rapide (IKr). Des mutations pathogènes dans ce gène qui réduit la fonction du canal Kv11.1 prolongent la durée de l’intervalle QT (Figure 1) et sont causales à LQTS2. Vingt-neuf pour cent des attaques syncopales dans LQTS2 se produisent pendant le repos / sommeil et seulement 13% des attaques syncopales ont été rapportées pendant l’exercice (25, 34). Des bruits surprenants soudains, par exemple des sonneries de réveil, des sonnettes de porte, des sonneries de téléphone, déclenchent généralement des crises syncopales chez ces patients (24, 34). Les patients présentant des mutations dans la région poreuse du Kv11.1 (codée par le gène KCNH2) sont sensibles à un risque élevé d’événements cardiaques liés à l’arythmie par rapport aux patients présentant des mutations dans la région non poreuse (35). Chez les nouveau-nés, le bloc auriculo-ventriculaire (AV) 2:1 est préférentiellement associé à des mutations KCNH2 (36). Un bloc AV complet compliqué par des LQTS a également été trouvé chez 17% des patients adultes présentant une mutation du gène KCNH2 (37). Les mutations homozygotes de KCNH2 sont rares et lorsqu’elles sont présentes, les patients souffrent d’une forme sévère de LQTS, avec 2:1 Bloc AV et arythmies ventriculaires sévères, pendant les stades intra-utérins ainsi qu’après la naissance (11, 38-40). En plus des nombreuses mutations décrites dans la pathologie LQTS2, des variants polymorphes courants dans le gène KCNH2 pourraient également moduler la gravité de la maladie. Un exemple intrigant est le polymorphisme du K897T (SNP) dans le KCNH2, qui est présent dans 33% de la population générale (41). Il a été rapporté que le K897T exacerbait la pathogénicité des mutations du KCNH2 (42, 43).
LQTS3
Nav1.5 est la sous-unité α formant des pores du canal Na + cardiaque voltage-dépendant, c’est une protéine membranaire intégrale codée par le gène SCN5A et est impliquée dans l’initiation et la conduction des potentiels d’action cardiaques. Les canaux Na+ cardiaques sont composés d’une sous-unité α formant des pores (codée par SCN5A) et d’une ou plusieurs sous-unités β auxiliaires.
LQTS3 est causé par un gain de mutations fonctionnelles qui perturbent l’inactivation rapide de la sous-unité α (Figure 1) et une mutation du gène SCN5A a été décrite chez < 10% de tous les patients atteints de LQTS avec une mutation (1, 20, 24). Les patients atteints de LQTS3 présentent la majorité (39%) de leurs événements cardiaques pendant le sommeil / repos (25), et environ113% des événements se sont produits pendant l’exercice (25). Dans plusieurs cas, il a été démontré qu’une seule mutation SCN5A exerce deux ou même trois phénotypes distincts d’arythmies dans la même famille, par exemple, LQTS, BrS ou CCD (44-47). Les patients masculins porteurs d’une mutation LQTS3 pourraient développer des symptômes beaucoup plus tôt que les patientes (48). Des mutations hétérozygotes et homozygotes du SCN5A ont été décrites dans LQTS3 avec un bloc AV fonctionnel 2:1 (49). Nous avons récemment trouvé une famille iranienne avec la mutation 1507_1509delQKP chez plusieurs membres de la famille, où les patients avaient combiné LQTS et CCD (données non montrées). Cette mutation a également été rapportée chez des patients d’autres pays, ce qui suggère qu’il s’agit d’une mutation récurrente et à point chaud. Des mutations présentant de telles caractéristiques de perte et de gain de fonction au cours de différentes phases du potentiel d’action ont également été rapportées dans les mutations 1493delK et 1795insD (44, 51).
Occasionnellement, un SNP pourrait également exercer un effet pathogène sur ses porteurs. S1103Y est une variante courante du gène SCN5A, présente chez 13% des Afro-Américains (52). Les porteurs de cette variante présentent un risque accru d’arythmies et de syndrome de mort subite du nourrisson (SMSN) (52).
LQTS4
LQTS4 représente la première forme non-canal de LQTS. Une mutation de l’ANK2, une protéine adaptatrice, entraîne une surcharge de calcium intracellulaire qui contribue au LQTS4 (53, 54). En plus de l’allongement de l’intervalle QT, les patients atteints de ce syndrome pourraient présenter une bradycardie sinusale, une FA paroxystique et une CPVT (54). L’effet pathogène des mutations ANK2 peut être modéré à sévère et les expressions cliniques dépendent de la gravité de la mutation.
LQTS5
Les mutations de KCNE1 sont associées au LQTS5 (Figure 1) (55, 56). Les mutations hétérozygotes du KCNE1 réduisent l’IKs en exerçant un effet négatif dominant sur l’allèle normal qui l’accompagne et entraînent un retard de la repolarisation cardiaque (Figure 1), responsable d’un risque accru d’arythmie (6). Les patients présentant des mutations homozygotes de KCNE1 souffrent de JLNS (type 2) (57, 58).
D85N est un polymorphisme du gène KCNE1, présent dans 0.7-1% de la population générale (41). Dans une étude de Nishio et al. (59), le polymorphisme du D85N a été plus fréquemment observé chez les patients atteints de LQTS, ce qui en fait un génotype à risque dans la pathologie du LQTS (potentiellement uniquement dans la population asiatique). En Europe, le D85N a été rapporté chez 5% des patients atteints de LQTS acquis (aLQTS) (dans une cohorte de 32 patients), qui avaient présenté un TdP (60).
LQTS6
Le gène KCNE2 code pour le peptide 1 lié au vison (MiRP1), une sous-unité β putative du canal potassique cardiaque IKr (Figure 1). Des mutations du gène KCNE2 pourraient également entraîner des défauts de la composante à activation rapide du courant potassique redresseur retardé (IKr), base pathologique du LQTS6 (61). Stimulus auditif / acoustique comme le bruit du réveil, la sonnette, etc. pourrait provoquer des attaques syncopales chez les porteurs de la mutation KCNE2, similaires à la mutation KCNH2 (62).
LQTS7
Ce syndrome est également connu sous le nom de syndrome d’Andersen–Tawil (ATS). L’ATS est un trouble rare, qui se manifeste par une syncope occasionnelle et un arrêt cardiaque. Les caractéristiques de l’ECG comprennent un léger allongement de l’intervalle QT, des ondes U anormales, une ectopie ventriculaire fréquente, une tachycardie ventriculaire bidirectionnelle (VT) et une VT polymorphe. Ce syndrome présente également des caractéristiques extracardiaques, par exemple une paralysie périodique du muscle squelettique et des problèmes de développement, tels qu’une fente palatine, des oreilles basses, une petite taille et des défauts de développement dans les membres (63). La majorité des patients atteints de STA diagnostiqués cliniquement présentaient une mutation du KCNJ2 (63). KCNJ2 code pour une sous-unité formant des pores de canaux potassiques redresseurs vers l’intérieur (IK1) (Figure 1) (64, 65).
LQTS8
Également connu sous le nom de syndrome de Timothy (TS), les patients présentent un allongement de l’intervalle QT sévère sur leur ECG, associé à une syndactylie, à une calvitie à la naissance et à de petites dents dans 100% des cas et à moins de malformations structurelles cardiaques pénétrantes, de retard mental, d’autisme et de caractéristiques dysmorphiques faciales (66). Il existe deux sous-types: TS1 (classique) et TS2 (forme rare).
TS2 est cardiologiquement plus sévère que TS1 (66, 67). Les patients TS2 manquent également de syndactylie (67). Des mutations dans la sous-unité α-1 du gène codant le courant calcique de type L (ICa-L) CACNA1C conduisent aux deux formes de TS (LQTS8). Il existe alternativement un exon-8 épissé et mutuellement exclusif dans le gène CACNA1C, pour plus de clarté, ils sont nommés exon-8 et exon-8A. Dans le cœur et le cerveau, où le CACNA1C est principalement exprimé, l’exon-8 se trouve dans ∼80% des transcrits d’ARNM et l’exon-8A est présent transc20% des transcrits (66). La mutation G406R dans l’exon-8A est causale de la forme classique de TS (TS1) et G402S dans l’exon-8 a été rapportée dans severer dans TS2. Un nouvel ajout à cette liste est la mutation Ala1473Gly, qui a été décrite chez un nourrisson TS avec un phénotype élargi (68). Toutes les mutations étaient soit de novo, soit mosaic chez le parent, et toutes résultaient d’un gain de fonction du canal ICa-L (66-69).Les mutations
LQTS9 et LQTS10
dans CAV3 ou SCN4B produisent un gain de fonction dans l’INa tardif, provoquant un phénotype de type LQTS3 (70-74). Ils sont connus sous le nom de LQTS9 (associé à la mutation CAV3) et LQTS10 (associé à la mutation SCN4B).
Les cavéoles sont bien décrites par Engelman et al. (72) comme de “petites grottes” dans la membrane plasmique. Ce sont de petites fosses non revêtues et sont considérées comme le site d’événements dynamiques et réglementaires importants au niveau de la membrane plasmique (72, 73). Les cavéolines sont les principales protéines des cavéoles, et la cavéoline-3 (codée par le gène CAV3) se trouve spécifiquement dans les cardiomyocytes et les cellules musculaires squelettiques. Plusieurs canaux ioniques cardiaques ont été spécifiquement localisés dans les cavéoles extraites de myocytes cardiaques enrichies en cavéoline-3 (72, 73). De plus, des composants de la cascade de signalisation des récepteurs β-adrénergiques sont également présents dans les membranes enrichies en cavéoles (72, 73).
SCN4B code pour NaVß4, qui est une sous-unité β auxiliaire du canal sodique cardiaque. Jusqu’à présent, une seule mutation (L179F) a été rapportée dans ce gène dans une famille mexicaine avec plusieurs membres de la famille affectés (74). La mutation de ce gène a entraîné un gain de fonction du courant Nav1.5 (74).
LQTS11
Dans le cœur, la régulation sympathique de la durée du potentiel d’action cardiaque (DPA) est médiée par l’activation du récepteur β-adrénergique (β-AR), qui nécessite l’assemblage d’AKAP9 (Yotiao) avec la sous-unité α (KvLQT1) du canal IKs. La mutation dans AKAP9 provoque LQTS11(75). À ce jour, une seule mutation, S1570L, chez AKAP9 a été rapportée (75).La mutation
LQTS12
du gène α-1-Syntrophine (SNTN1) est causale de LQTS12. La mutation de ce gène conduit à un gain de fonction du canal sodique cardiaque (Nav1.5), qui est la base pathologique du LQTS12 (76).
LQTS13
La sous-unité de canal potassique rectifiant vers l’intérieur couplée aux protéines G (Kir3.4) est codée par le gène KCNJ5. Une mutation de perte de fonction dans ce gène pourrait provoquer LQTS13(77). Jusqu’à présent, une seule mutation, G387R, a été décrite dans une famille chinoise avec neuf patients porteurs de cette mutation. Une expression membranaire plasmatique réduite de Kir3.4 a été suggérée comme pathologie des LQTS chez les patients.
LQTS acquis
En plus des LQTS congénitaux, il existe également une autre variante des LQTS connue sous le nom d’ALQTS, qui est causée par des facteurs et des substances qui diminuent le flux de potassium et altèrent la capacité du myocarde à se repolariser. Les affections bien connues sont le sexe féminin, l’hypokaliémie et les médicaments qui inhibent les canaux potassiques cardiaques (78-80). Un certain nombre de médicaments couramment prescrits pourraient également se lier et bloquer préférentiellement le canal HERG (Kv11.1, une protéine codée par le gène KCNH2) et prédispose aux ALQTS (79, 80). Récemment, il a été démontré que le blocage des IKS pouvait également contribuer aux ALQTS induits par la drogue, en particulier lorsque la réserve de repolarisation est compromise (81). Il a été constaté que la fluoxétine et la norfluoxétine supprimaient les propriétés de l’IKs, à la fois in vivo et in vitro, et ont conduit à des LQTS marqués (81). Des polymorphismes, D85N chez le vison (gène KCNE1), T8A, Q9E chez MiRP1 (gène KCNE2), qui sont des sous-unités β putatives des canaux IKs et IKr, ont été rapportés pour provoquer des aLQTS (82, 83). Des LQTS auto-immunes ont également été rapportées chez un patient présentant des IgG contenant des anticorps anti-HERG (84).
Caractéristiques électrocardiographiques dans les Trois formes courantes du syndrome du QT long
Des modèles typiques d’ondes ST-T sont présents chez la majorité des patients atteints de LQTS génotypés et peuvent être utilisés pour identifier les génotypes LQTS1, LQTS2 et éventuellement LQTS3 (85, 86).
La forme LQTS1 du LQTS est associée à une onde T large sans raccourcissement de l’intervalle QT à l’exercice (Figure 2A). LQTS2 est associé à des ondes T de faible amplitude, souvent bifides (Figure 2B). LQTS3 est associé à un segment iso-électrique long et à une onde T haute à base étroite (figure 2C). La dépendance à la pause de l’apparition du TdP dans les TQT congénitales est spécifique au génotype, étant prédominante dans les TQT2, mais presque absente dans les TQT1 (87).

Figure 2. Enregistrements d’électrocardiogrammes de patients atteints de LQTS1, LQTS2 et LQTS3. (A) ECG à 12 dérivations d’un homme de 18 ans avec une mutation KCNQ1. L’intervalle QT est prolongé (QTc = ±500 ms). Le segment ST a une base large et une amplitude relativement grande. L’intervalle de conduction est normal (étalonnage standard). (B) ECG à 12 dérivations d’une fille de 14 ans avec une mutation KCNH2. L’intervalle QT est prolongé (QTc ± 520 ms). Le segment ST est entaillé dans la sonde V3 et présente une amplitude relativement faible dans les sondes d’extrémité. L’intervalle de conduction est normal (étalonnage standard). (C) ECG à 12 dérivations d’un garçon de 12 ans avec une mutation SCN5A. L’intervalle QT est prolongé (QTc ± 600 ms). Le segment ST a un long segment (presque) iso-électrique avec une onde T grande, forte et étroite. L’intervalle de conduction est normal (étalonnage standard).
Bien que les modèles puissent suggérer un génotype spécifique du LQTS, des exceptions ont été fréquemment trouvées.
Génotype-Phénotype
Le syndrome du QT long est une maladie autosomique dominante. L’analyse du génotype et du phénotype chez les porteurs de mutations hétérozygotes a été menée assez largement chez les patients LQTS1, LQTS2 et LQTS3. Pendant l’enfance, le risque d’événements cardiaques est significativement plus élevé chez les hommes LQTS1 que chez les femmes LQTS1, alors qu’aucune différence significative de risque d’événements cardiaques liée au sexe n’a été observée chez les patients LQTS2 et LQTS3 (88-91). À l’âge adulte (également après l’âge de 40 ans), les femelles LQTS1 et LQTS2 pourraient présenter un risque significativement plus élevé d’événements cardiaques que les mâles respectifs (88-91). En général, la létalité des événements cardiaques semble être prédominante chez les patients LQTS3 que chez les patients LQTS1 et LQTS2 (91). Les femmes atteintes de LQTS ont un risque réduit d’événements cardiaques pendant la grossesse, mais le risque augmente tout à fait pendant la période post-partum de 9 mois, en particulier chez les femmes présentant une mutation du gène KCNH2 (92).
La mort cardiaque subite chez les enfants pourrait également être causée par des mutations dans les gènes du canal ionique cardiaque (93-96). Environ 28% des enfants atteints d’un SCD inexpliqué (entre 1 et 18 ans, âge moyen: 12,3 ± 3,8 ans) étaient porteurs de mutations dans les gènes causaux du LQTS (97). Chez les SMSN, les mutations de SCN5A semblaient prédominantes (98, 99), mais des mutations de KCNQ1, KCNH2, KCNE2 et CAV3, SCN4B et SCN3B ont également été trouvées (100, 101). Des décès fœtaux intra-utérins ont également été signalés en raison de défauts dans les gènes des canaux ioniques cardiaques (12, 102).
La prise en charge clinique des LQTS
L’arrêt de tous les médicaments connus pour prolonger l’intervalle QT ainsi que la correction des déséquilibres électrolytiques et / ou des conditions métaboliques précipitantes devrait être l’objectif principal lors du traitement des patients (acquis) LQTS. Les symptômes dans les TQL sont souvent médiés par les récepteurs adrénergiques, par conséquent, une restriction de la participation des patients aux activités sportives est généralement recommandée (24, 25). Le pilier de la thérapie clinique pour le LQTS est le β-blocage. Des préparations à action prolongée de propranolol, de nadolol et de métoprolol sont généralement utilisées, et leur efficacité dans le blocage β est évaluée par émoussement de la fréquence cardiaque d’exercice (par exemple, par >20%) (24, 25). Parmi tous les β-bloquants, le propranolol et le nadolol sont considérés comme supérieurs au métoprolol chez les patients symptomatiques (103). En outre, le blocage β peut également être utilisé comme traitement prophylactique chez les porteurs de mutations silencieuses pour réduire le SCD (24). Dans une étude de Barsheshet et al. (30), les patients présentant des mutations faux sens de la boucle C dans le gène KCNQ1 présentaient un risque élevé d’événements cardiaques mettant leur vie en danger et ils avaient des avantages significatifs du traitement par β-bloquants. Comme les femmes atteintes de LQTS2 présentent un risque accru au cours de la période post-partum de 9 mois, des β-bloquants doivent être prescrits pour réduire tout événement cardiaque au cours de cette période à haut risque (92). Un défibrillateur–cardioverteur implantable (DCI) peut être envisagé chez les patients présentant une syncope récurrente malgré un traitement par β-bloqueur ou chez les patients présentant un risque élevé d’arrêt cardiaque (par ex., LQTS2 symptomatiques et LQTS3 avec un allongement de l’intervalle QTc documenté). La dénervation sympathique cardiaque gauche (LCSD) est recommandée chez les patients atteints de TQL à haut risque chez lesquels un DCI est contre-indiqué ou refusé et où les β-bloquants ne sont pas efficaces, non tolérés, non acceptés ou contre-indiqués (18, 104). Les patients atteints de JLNS ont généralement un QTc > 500 ms et sont également à haut risque, les β-bloquants ont une efficacité insuffisante chez ces patients chez lesquels un traitement précoce par CIM a été recommandé (18, 31).
LQTS: Perspective saoudienne
Le premier rapport sur les QQT en Arabie saoudite a été publié en 1993 à l’Hôpital des Forces armées de Riyad (105). Quatre nourrissons et jeunes enfants âgés de 6 à 48 mois ayant des antécédents de crises récurrentes, issus d’une seule famille, ont été diagnostiqués comme des LQTS (105). Les antécédents familiaux ont révélé que deux autres membres de la famille élargie avaient des épisodes similaires de perte soudaine de conscience et que trois membres de la famille sont décédés subitement (105). Dans tous les cas, le diagnostic initial était l’épilepsie (105). Plusieurs années plus tard, deux rapports de cas sporadiques avec une variante relativement plus sévère du LQTS néonatal associée à un bloc AV 2: 1 ont été rapportés (106, 107). Tous ces rapports cliniques publiés étaient sans résultats génétiques pouvant expliquer la physiopathologie des LQTS chez eux (105-107).
Pour la première fois, nous avons signalé des anomalies génétiques comme base pathologique de LQTS chez des patients similaires d’Arabie saoudite. Nous avons enquêté sur six familles saoudiennes ayant des antécédents de syncope et de décès inexpliqués soudains de fœtus, de nouveau-nés et d’enfants (12-14 ans). Le LQTS1 autosomique récessif a été diagnostiqué chez des enfants de deux familles (figure 3). Le LQTS2 autosomique récessif a été diagnostiqué dans deux familles (figure 4). Dans une famille, une patiente a reçu un diagnostic de LQTS2 autosomique dominante (Figure 5), la patiente a eu des crises syncopales pendant la période de récupération post-partum à l’hôpital, ce qui est très fréquent chez les femmes porteuses de la mutation KCNH2. Chez tous nos patients, l’identification de mutations pathogènes dans les gènes du canal ionique cardiaque causal LQTS a conduit à un diagnostic clinique confirmé, qui ont été mal diagnostiqués comme des convulsions épileptiques avant de nous être référés (13, 14) Récemment, Shinwari et al. (15) de l’Hôpital spécialisé King Faisal et du Centre de recherche ont signalé une mutation causale du KCNQ1 LQTS1, H258P, dans une famille nombreuse comptant 12 personnes touchées. Seuls deux porteurs étaient symptomatiques, leur QTc était > 500 ms, et les β-bloquants supprimaient les symptômes cliniques chez un patient et le second patient symptomatique nécessitait un DCI.
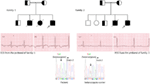
Figure 3. Dessin de Pedigree de la famille – 1 et 2 avec LQTS1 autosomique récessif, les probands sont représentés par une flèche. Les ECG des probands de deux familles sont représentés au milieu, avec un QTc de 557 et 529 ms, respectivement. Une mutation intronique, c. 387-5 T > A dans le gène KCNQ1 a été trouvée chez les patients (indiquée en bas). La mutation a été montrée avec une flèche. Les cercles et les carrés non remplis ne sont pas porteurs de la mutation. Les individus affectés sont représentés sous forme de cercles remplis (femelle) et de carrés (mâle). Les carrés et les cercles à moitié remplis sont des individus présentant une mutation hétérozygote. Les individus décédés sont indiqués par des barres obliques, les probands sont indiqués par une flèche et le mariage consanguin est indiqué par = La limite Exon−intron est indiquée par une ligne pointillée avec une flèche pointant vers l’exon.
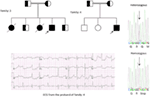
Figure 4. Dessin de Pedigree de la famille – 3 et 4 avec LQTS2 autosomique récessif. L’ECG du proband de la famille-4 est montré sous le dessin de pedigree, qui montre une tachycardie sinusale, un bloc AV presque complet, un rythme d’échappement complexe large avec de très longs intervalles QTc (QT > 600 ms). Une mutation, c. 3208 C > T (p. Q1070X) dans le gène KCNH2 a été trouvée chez les patients (montré à droite), marquée d’une flèche.
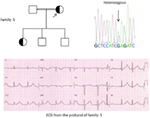
Figure 5. En haut à gauche : pedigree de la famille – 5. L’individu affecté est représenté par un cercle rempli (femelle) et est indiqué par une flèche. ECG à 12 dérivations du proband (I:2). L’ECG montre une onde T haute, large et de grande amplitude, l’intervalle QTc est de 580 ms. Le dépistage du gène KCNH2 montre une substitution du nucléotide “G” par un “A” (c. 2362G > A, flèche marquée), ce qui conduit à une substitution d’acides aminés, p. E788K.
Les résultats génétiques et cliniques de notre étude (12-14) en Arabie saoudite sont assez intrigants pour plusieurs raisons: (1) Au total, nous avons étudié six familles, dont quatre étaient homozygotes / hétérozygotes composés pour les mutations et les mutations provenaient d’une source ancestrale; (2) Toutes les mutations dans les LQTS étaient nouvelles, rapportées uniquement dans ces familles arabes; (3) En raison de l’homozygotie ou de l’hétérozygotie composée pour les mutations, les phénotypes cliniques étaient également sévères dans nos familles étudiées (12-14). Nous avons suggéré que les observations génétiques et phénotypiques découlaient du taux extrêmement élevé de mariages consanguins en Arabie saoudite (108, 109). Notre étude a fourni les premières preuves scientifiques sur le rôle de la consanguinité dans l’exercice d’un rôle pivot dans les arythmies cardiaques inexpliquées et les SCD chez les enfants et les adolescents en Arabie saoudite (12-14). Nous avons également montré que la mutation KCNQ1 c. 387 -5 T > A (NM_000218) (Figure 3) s’était propagée dans la province d’Assir en Arabie saoudite à partir d’un ancêtre commun pendant plusieurs générations en raison de la forte incidence des mariages consanguins. Jusqu’à présent, il s’agit de la mutation causale LQTS1 la plus courante dans la population saoudienne, qui a également été observée à l’Hôpital spécialisé King Faisal et à l’Hôpital militaire Khamis Mashayt (non publié). Un résultat similaire a également été obtenu pour la mutation causale de LQTS2, c. 3208 C > T (p.Q1070X) (Figure 4) dans le gène KCNH2 (12, 14), qui est également une mutation fondatrice en Arabie saoudite et séparée pendant de nombreuses générations dans la région d’Assir (voir la carte, Figure 6). Nous prédisons qu’il existe un nombre considérable d’individus avec les mutations ancestrales / fondatrices mentionnées (à la fois dans KCNQ1 et KCNH2 et d’autres gènes) dans cette région et aussi dans les grandes villes comme Riyad, Jeddah et Dammam en raison de la migration urbaine. D’autres mutations fondatrices ou ancestrales pathogènes pour les LQTS sont très envisagées dans d’autres provinces d’Arabie saoudite en raison du taux élevé de mariages consanguins.
Figure 6. Carte de l’Arabie saoudite (avec la permission de Wikipedia). La région d’Assir est marquée d’une ligne épaisse, où nous avons trouvé les mutations fondatrices dans les gènes KCNQ1 et KCNH2, décrits dans la famille 1-4.
Comme la LQTS est une maladie autosomique dominante, nous nous attendions à observer des patients porteurs de mutations principalement hétérozygotes. Mais dans notre étude, nous avons principalement identifié des patients présentant des mutations récessives (12-14). De toute évidence, ces patients deviennent d’abord symptomatiques et ont été principalement dirigés vers le Centre cardiaque Prince Sultan, les portant sous notre attention. Néanmoins, les résultats de nos enquêtes impliquent le fait que les LQTS récessifs pourraient avoir des phénotypes cliniques mortels chez les enfants, et ils ne sont pas rares en Arabie saoudite (12-14). Comme les porteurs de mutations hétérozygotes sont également susceptibles de développer une arythmie et ses complications, une initiative concertée devrait être prise pour réunir les médecins généralistes, les cardiologues et les généticiens cliniques locaux sur une plate-forme commune pour identifier les personnes à risque. De plus, pour le moment, nous n’avons pas non plus d’informations génétiques pour d’autres troubles de l’arythmie familiale, par exemple, CPVT, SQTS, BrS, AF, etc. Les centres cardiogénétiques spécialisés devraient prendre l’initiative de rechercher les défauts génétiques, les mutations et effectuer des études génotype-phénotype dans toutes les formes d’arythmies héréditaires. Il convient également de garder à l’esprit que toutes les arythmies génétiques n’auraient pas d’antécédents familiaux, car dans de nombreux cas, la mutation causale de l’arythmie est d’origine novo, c’est-à-dire que le proband est le premier patient de cette famille avec la mutation et qu’il est la source de transmission de la mutation dans les générations en aval (110, 111). En raison du taux très élevé de mariages consanguins en Arabie saoudite, nous nous attendons à ce que beaucoup plus de mutations fondatrices exercent un rôle crucial dans les arythmies congénitales dans ce pays. L’identification de ces mutations fondatrices devrait être notre tâche première, ce qui nous faciliterait le développement d’un conseil génétique prénuptial et pré-symptomatique efficace dans ce pays. Des mutations dans les gènes causaux du LQTS pourraient conférer une variabilité de la pénétrance clinique, à un extrême, certains porteurs pourraient être vraisemblablement complètement sains, mais certains porteurs pourraient avoir leur première manifestation de la maladie sous forme de syncope ou de mort subite. La mort subite d’un jeune enfant ou d’un adulte pose un lourd fardeau psychologique et émotionnel à la famille, le dépistage des porteurs est essentiel car il existe des médicaments simples, par exemple des β-bloquants (également modification du comportement), qui pourraient très efficacement prévenir les porteurs des conséquences fatales de l’arythmie et des SCDs. Les personnes présentant des mutations homozygotes dans les gènes causaux du LQTS pourraient présenter une morbidité sévère et un taux de mortalité extrêmement élevé, y compris des tachyarythmies de brady fœtales et, dans de nombreux cas, des fausses couches, des avortements spontanés chez les femmes enceintes (12-14).
Peu de recherches ont également été menées en Arabie saoudite sur la prévalence de divers SNP dans les gènes liés à l’arythmie et également dans les gènes qui les régulent. Splawski et coll. (112) ont décrit un variant S1103Y commun dans le gène SCN5A associé à une arythmie chez les Afro-Américains. L’allèle variant appelé Y1103 est responsable de l’accélération de l’activation du canal, augmentant ainsi la probabilité d’arythmies cardiaques chez les personnes d’ascendance africaine (52, 112). K393N est un variant du gène KCNQ1 rapporté chez des patients LQTS1 aux États-Unis, mais, dans la population arabe, nous avons détecté ce variant chez 2% des individus (données non publiées). La question de savoir si la variante K393N du gène KCNQ1 chez les Arabes est comparable à la variante S1103Y (SCN5A) chez les Afro-Américains ou la variante D85N (KCNE1) dans la population japonaise pourrait également mériter une enquête (59).
Déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de relations commerciales ou financières pouvant être interprétées comme un conflit d’intérêts potentiel.
Remerciements
Notre sincère gratitude au Prof. Arthur Wilde, au Prof. Connie Bezzina et à l’éditeur de la revue “Heart” pour leur permission d’utiliser la Figure 1 dans ce manuscrit de Réf. (113).
1. Bhuiyan ZA. Spectre clinique et génétique des Syndromes d’Arythmie cardiaque Héréditaires. Amsterdam : Université d’Amsterdam (2009).
2. A priori SG, Aliot E, Blømstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, et al. Groupe de travail sur la mort subite cardiaque, Société européenne de cardiologie. Europace (2002) 4:3-18. doi: 10.1053 / eupc.2001.0214
CrossRef Texte intégral
3. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL et coll. Mutations SCN5A associées à une arythmie cardiaque héréditaire, syndrome du QT long. Cell (1995) 80:805-11. doi:10.1016/0092-8674(95)90359-3
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
4. Il s’agit de l’un des plus grands groupes de la planète. Une base moléculaire pour l’arythmie cardiaque: les mutations de HERG provoquent un syndrome du QT long. Cell (1995) 80:795-803. doi: 10.1016/0092-8674(95)90358-5
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
5. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, et al. Clonage positionnel d’un nouveau gène du canal potassique : les mutations KVLQT1 provoquent des arythmies cardiaques. Nat Genet (1996) 12:17-23. doi: 10.1038/ng0196-17
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
6. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Les mutations du gène hminK provoquent un syndrome du QT long et suppriment la fonction IKs. Nat Genet (1997) 17:338-40. doi: 10.1038/ng1197-338
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
7. Sanguinetti MC, Jiang C, Curran ME, Keating MT. Un lien mécaniste entre une arythmie cardiaque héréditaire et une arythmie cardiaque acquise : HERG code le canal potassique IKr. Cell (1995) 81:299-307. doi:10.1016/0092-8674(95)90340-2
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. Une nouvelle mutation dans le gène du canal potassique KVLQT1 provoque le syndrome cardioauditaire de Jervell et de Lange-Nielsen. Nat Genet (1997) 15:186-9. doi: 10.1038/ng0297-186
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
9. Kucera JP, Rohr S, Rudy Y. La localisation des canaux sodiques dans les disques intercalés module la conduction cardiaque. Circ Res (2002) 91:1176-82. doi: 10.1161/01.RÉS.0000046237.54156.0A
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
10. Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Connexin43 remodelage causé par l’inhibition de l’expression de la plakophiline-2 dans les cellules cardiaques. Circ Res (2007) 101:703-11. doi: 10.1161/ CIRCRESAHA.107.154252
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
11. Rohr S. Diaphonie moléculaire entre les jonctions mécaniques et électriques au niveau du disque intercalé. Circ Res (2007) 101:637-9. doi: 10.1161/ CIRCRESAHA.107.161901
CrossRef Texte intégral
12. Il s’agit de l’un des plus grands groupes de la planète. Perte fœtale intra-utérine récurrente due à une quasi-absence de HERG: caractérisation clinique et fonctionnelle d’une mutation homozygote non-sens HERG Q1070X. Rythme cardiaque (2008) 5:553-61. doi: 10.1016/j. hrthm.2008.01.020
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
13. Il s’agit de l’une des principales sources d’information sur la santé et la sécurité au travail. Une mutation intronique conduisant à un saut incomplet de l’exon-2 dans KCNQ1 sauve l’audition dans le syndrome de Jervell et de Lange-Nielsen. Prog Biophys Mol Biol (2008) 98:319-27. doi: 10.1016 / j. pbiomolbio.2008.10.004
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
14. Bhuiyan ZA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi S, Al-Kalaf K, Mannens MMAM, et al. Analyse clinique et génétique du syndrome du QT long chez les enfants de six familles en Arabie saoudite: sont-ils différents? Pediatr Cardiol (2009) 30:490-501. doi: 10.1007/s00246-008-9377- y
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
15. Shinwari ZM, Al-Hazzani A, Dzimiri N, Tulbah S, Mallawi Y, Al-Fayyadh M, et al. Identification d’une nouvelle mutation KCNQ1 dans une grande famille saoudienne atteinte du syndrome du QT long: conséquences cliniques et implications préventives. Clin Genet (2013) 83:370-4. doi: 10.1111/j.1399-0004.2012.01914.x
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prévalence du syndrome congénital du QT long. Circulation (2009) 120:1761-7. doi: 10.1161 / CIRCULATIONAHA.109.863209
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
17. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Critères diagnostiques pour le syndrome du QT long. Une mise à jour. Circulation (1993) 88:782-4. doi: 10.1161/01.CIR.88.2.782
CrossRef Texte intégral
18. A priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. Résumé: Déclaration de consensus HRS / EHRA / APHRS sur le diagnostic et la prise en charge des patients atteints de syndromes d’arythmie primaire héréditaire. Europace (2013) 15:1389-406. doi: 10.1093/europace/eut272
CrossRef Texte intégral
19. Liu JF, Jons C, Moss AJ, McNitt S, Peterson DR, Qi M, et al. Registre du syndrome. Facteurs de risque de syncope récurrente et d’événements mortels ou quasi mortels subséquents chez les enfants et les adolescents atteints du syndrome du QT long. Je suis Coll Cardiol (2011) 57:941-50. doi: 10.1016 / j.jacc.2010.10.025
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
20. Il s’agit de l’un des principaux facteurs de risque de la maladie d’alzheimer et de la maladie de Parkinson. Spectre de mutations dans les gènes du syndrome du QT long. KCNE1 et KCNE2. Circulation (2000) 102:1178-85. doi: 10.1161/01.CIR.102.10.1178
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
21. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Mutations composées: une cause fréquente de syndrome sévère du QT long. Circulation (2004) 109: 1834-41. doi: 10.1161/01.CIR.0000125524.34234.13
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
22. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, et al. Risque d’événements cardiaques mettant la vie en danger chez les patients atteints du syndrome du QT long et de mutations multiples. Rythme cardiaque (2013) 10:378-82. doi: 10.1016/j. hrthm.2012.11.006
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
23. Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Tests génétiques dans le syndrome du QT long: développement et validation d’une approche efficace du génotypage en pratique clinique. JAMA (2005) 294:2975-80. doi: 10.1001/ jama.294.23.2975
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
24. Roden DM. Pratique clinique. Syndrome du QT long. (2008) 358:169-76. doi: 10.1056/NEJMcp0706513
CrossRef Texte intégral
25. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Corrélation génotype-phénotype dans le syndrome du QT long: déclencheurs spécifiques aux gènes des arythmies potentiellement mortelles. Circulation (2001) 103:89-95. doi: 10.1161/01.CIR.103.1.89
CrossRef Texte intégral
26. Ackerman MJ, Testeur DJ, Portier CJ. La natation, un déclencheur arythmogène spécifique au gène du syndrome du QT long héréditaire. Mayo Clin Proc (1999) 74:1088-94. doi:10.4065/74.11.1088
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
27. Kapa S, Testeur DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, et al. Tests génétiques pour le syndrome du QT long: distinguer les mutations pathogènes des variantes bénignes. Circulation (2009) 120:1752-60. doi: 10.1161 / CIRCULATIONAHA.109.863076
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
28. Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, et al. Aspects cliniques du syndrome du QT long de type 1 par localisation, type de codage et fonction biophysique des mutations impliquant le gène KCNQ1. Circulation (2007) 115:2481-9. doi: 10.1161 / CIRCULATIONAHA.106.665406
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
29. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. Des variants dans la région 3′ non traduite du canal potassique Kv7.1 codé par KCNQ1 modifient la gravité de la maladie chez les patients atteints du syndrome du QT long de type 1 de manière spécifique à l’allèle. Cœur Eur J (2012) 33:714-23. doi: 10.1093/eurheartj/ehr473
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
30. Barsheshet A, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu W, et al. Mutations dans les boucles cytoplasmiques du canal KCNQ1 et risque d’événements potentiellement mortels: implications for mutation-specific response to β-blocker therapy in type 1 long-QT syndrome. Circulation (2012) 125: 1988-96. doi: 10.1161 / CIRCULATIONAHA.111.048041
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
31. Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. Le syndrome de Jervell et Lange-Nielsen: histoire naturelle, base moléculaire et résultat clinique. Circulation (2006) 113:783-90. doi: 10.1161 / CIRCULATIONAHA.105.592899
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
32. Bhuiyan ZA, Wilde AA. IKs dans le cœur et l’ouïe, l’oreille peut faire avec moins que le cœur. Circ Cardiovasc Genet (2013) 6:141-3. doi: 10.1161/ CIRCGÉNÉTIQUE.113.000143
CrossRef Texte intégral
33. Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Mécanismes physiopathologiques des mutations du canal K+ KVLQT1 dominantes et récessives trouvées dans les arythmies cardiaques héréditaires. Hum Mol Genet (1997) 6:1943-9. doi: 10.1093/hmg/6.11.1943
CrossRef Texte intégral
34. Wilde AA, Jongbloed RJ, Doevendans PA, Düren DR, Hauer RN, van Langen IM, et al. Les stimuli auditifs comme déclencheur d’événements arythmiques différencient les patients liés à HERG (LQTS2) des patients liés à KVLQT1 (LQTS1). J Am Coll Cardiol (1999) 33:327-32. doi: 10.1016/S0735-1097(98)00578-6
CrossRef Texte Intégral
35. Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, et al. Risque accru d’événements arythmiques dans le syndrome du QT long avec des mutations dans la région des pores du canal potassique du gène lié à l’éther-a-go-go humain. Circulation (2002) 105:794-9. doi: 10.1161/hc0702.105124
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
36. Lupoglazoff JM, Denjoy I, Villain E, Fressart V, Simon F, Bozio A, et al. Syndrome du QT long chez les nouveau-nés: troubles de la conduction associés aux mutations HERG et bradycardie sinusale avec mutations KCNQ1. J Am Coll Cardiol (2004) 43:826-30. doi: 10.1016 / j.jacc.2003.09.049
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
37. Chevalier P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ, et al. Torsades de pointes compliquant le bloc auriculo-ventriculaire: preuve d’une prédisposition génétique. Rythme cardiaque (2007) 4:170-4. doi: 10.1016/j. hrthm.2006.10.004
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
38. Hoorntje T, Alders M, van Tintelen P, van der Lip K, Sreeram N, van der Wal A, et al. Troncation prématurée homozygote de la protéine HERG: le ko de HERG humain. Circulation (1999) 100:1264-7. doi: 10.1161/01.CIR.100.12.1264
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
39. Piippo K, Laitinen P, Swan H, Toivonen L, Viitasalo M, Pasternack M, et al. L’homozygotie pour une mutation des canaux potassiques de HERG provoque une forme sévère de syndrome du QT long : identification d’une mutation fondatrice apparente chez les Finlandais. J Am Coll Cardiol (2000) 35:1919-25. doi: 10.1016/S0735-1097(00)00636-7
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
40. Johnson WH Jr, Yang P, Yang T, Lau YR, Mostella BA, Wolff DJ, et al. Caractérisation clinique, génétique et biophysique d’une mutation homozygote de HERG provoquant un syndrome néonatal sévère du QT long. Pediatr Res (2003) 53:744-8. doi: 10.1203/01.PDR.0000059750.17002.B6
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
41. Ackerman MJ, Testeur DJ, Jones GS, Will ML, Burrow CR, Curran MOI. Différences ethniques dans les variantes des canaux potassiques cardiaques: implications for genetic susceptibility to sudden cardiac death and genetic testing for congénital long QT syndrome. Mayo Clin Proc (2003) 78:1479-87. doi:10.4065/78.12.1479
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
42. Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T est un modificateur génétique du syndrome congénital latent du QT long. Circulation (2005) 112:1251-8. doi: 10.1161 / CIRCULATIONAHA.105.549071
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
43. Nof E, Cordeiro JM, Pérez GJ, Scornik FS, Calloe K, Love B, et al. Un polymorphisme nucléotidique commun peut exacerber le syndrome de type 2 à QT long, entraînant la mort subite du nourrisson. Circ Cardiovasc Genet (2010) 3:199-206. doi: 10.1161/ CIRCGÉNÉTIQUE.109.898569
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
44. Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, et al. Une seule mutation du canal Na (+) provoquant à la fois des syndromes à QT long et des syndromes de Brugada. Circ Res (1999) 85:1206-13. doi: 10.1161/01.RES.85.12.1206
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
45. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, et al. Nouvelle mutation SCN5A conduisant soit à un défaut de conduction cardiaque isolé, soit au syndrome de Brugada dans une grande famille française. Circulation (2001) 104:3081-6. doi: 10.1161/hc5001.100834
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
46. Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, et al. La mutation E1784K dans le SCN5A est associée à un phénotype clinique mixte du syndrome du QT long de type 3. J Clin Invest (2008) 118:2219-29. doi: 10.1172/ JCI34057
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
47. Keller DI, Acharfi S, Delacrétaz E, Benammar N, Rotter M, Pfammatter JP, et al. Une nouvelle mutation dans SCN5A, delQKP 1507-1509, provoquant le syndrome du QT long: rôle du résidu Q1507 dans l’inactivation des canaux sodiques. J Mol Cell Cardiol (2003) 35:1513-21. doi: 10.1016/j. yjmcc.2003.08.007
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
48. A priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Stratification du risque dans le syndrome du QT long. (2003) 348:1866-74. doi: 10.1056/NEJMoa022147
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
49. Lupoglazoff JM, Cheav T, Baroudi G, Berthet M, Denjoy I, Cauchemez B, et al. Mutation SCN5A homozygote dans le syndrome du QT long avec bloc auriculo-ventriculaire fonctionnel de deux à un. Circ Res (2001) 89: E16–21. doi: 10.1161/hh1401.095087
CrossRef Texte intégral
50. Shi R, Zhang Y, Yang C, Huang C, Zhou X, Qiang H, et al. La mutation du canal sodique cardiaque delQKP 1507-1509 est associée au spectre phénotypique en expansion de la LQT3, à un trouble de la conduction, à une cardiomyopathie dilatée et à une incidence élevée de mort subite chez les jeunes. Europace (2008) 10:1329-35. doi: 10.1093/europace/eun202
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
51. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartscheer A, Eckardt L, Paul M, et al. Une mutation de délétion hétérozygote dans le gène du canal sodique cardiaque SCN5A avec des caractéristiques de perte et de gain de fonction se manifeste par une maladie de conduction isolée, sans signes de Brugada ou de syndrome du QT long. Il s’agit de la première version de PLoS One (2013) 8 : e67963. doi: 10.1371 / journal.pone.0067963
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
52. L’usine LD, Bowers PN, Liu Q, Morgan T, Zhang T, State MW, et al. Une variante du canal sodique cardiaque commune associée à la mort subite du nourrisson chez les Afro-Américains, SCN5A S1103Y.J Clin Invest (2006) 116:430-5. doi: 10.1172 / JCI25618
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
53. Il s’agit de l’un des principaux facteurs de risque de la maladie d’alzheimer et de la maladie de Parkinson. La mutation Ankyrine-B provoque une arythmie cardiaque de type 4 à QT long et une mort cardiaque subite. Nature (2003) 421:634-9. doi: 10.1038 /nature01335
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
54. Les juges Schott, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB et coll. Cartographie d’un gène du syndrome du QT long sur le chromosome 4q25-27. Am J Hum Genet (1995) 57:1114-22.
Résumé Pubmed / Texte intégral Pubmed
55. Les protéines Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K (V) LQT1 et lsK (minK) s’associent pour former le courant potassique cardiaque I (Ks). Nature (1996) 384:78-80. doi: 10.1038|384078a0
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
56. Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Coassemblage des protéines K(V) LQT1 et minK (IsK) pour former un canal potassique cardiaque I (Ks). Nature (1996) 384:80-3. doi: 10.1038 / 384080a0
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
57. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al. Les mutations KCNE1 provoquent le syndrome de Jervell et de Lange-Nielsen. Nat Genet (1997) 17:267-8. doi: 10.1038/ng1197-267
CrossRef Texte intégral
58. Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutation du gène de l’IKs associée aux formes de Jervell et de Lange-Nielsen et de Romano-Ward du syndrome du QT long. Circulation (1998) 97:142-6. doi: 10.1186/1471-2350-9-24
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85N, un polymorphisme KCNE1, est une variante du gène responsable de la maladie dans le syndrome du QT long. Je suis Coll Cardiol (2009) 54:812-9. doi: 10.1016 / j.jacc.2009.06.005
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
60. Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, et al. Variations génétiques de KCNQ1, KCNH2, SCN5A, KCNE1 et KCNE2 chez les patients atteints du syndrome du QT long induit par le médicament. Mol Med (2004) 82:182-8. doi: 10.1007/s00109-003-0522- z
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
61. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 forme des canaux potassiques IKr avec HERG et est associé à une arythmie cardiaque. Cell (1999) 97:175-87. doi: 10.1016/S0092-8674 (00) 80728-X
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
62. Gordon E, Panaghie G, Deng L, Bee KJ, Roepke TK, Krogh-Madsen T, et al. Une mutation KCNE2 chez un patient présentant une arythmie cardiaque induite par des stimuli auditifs et un déséquilibre électrolytique sérique. Cardiovasc Res (2008) 77:98-106. doi: 10.1093/cvr/cvm030
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
63. Plaster NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, et al. Les mutations de Kir2.1 provoquent les phénotypes électriques développementaux et épisodiques du syndrome d’Andersen. Cell (2001) 105:511-9. doi: 10.1016 / L0092-8674(01)00342-7
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Structure primaire et expression fonctionnelle d’un canal potassique redresseur vers l’intérieur de la souris. Nature (1993) 362:127-33. doi: 10.1038/362127a0
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
65. Raab-Graham KF, Radeke CM, Vandenberg CA. Clonage moléculaire et expression d’un canal potassique redresseur vers l’intérieur du cœur humain. Neuroreport (1994) 5:2501-5. doi: 10.1097/00001756-199412000-00024
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
66. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 le dysfonctionnement des canaux calciques provoque un trouble multisystémique incluant l’arythmie et l’autisme. Cell (2004) 119:19-31. doi: 10.1016/j. cell.2004.09.011
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
67. Spawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Trouble d’arythmie sévère causé par des mutations cardiaques des canaux calciques de type L. Proc Natl Acad Sci U S A (2005) 102:8089-96. doi: 10.1073/pnas.0502506102
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
68. Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L, et al. QT long, syndactylie, contractures articulaires, accident vasculaire cérébral et nouvelle mutation CACNA1C: élargir le spectre du syndrome de Timothy. Am J Med Genet A (2012) 158A: 182-7. doi: 10.1002/ ajmg.a.34355
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
69. Dufendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. Le mosaïcisme maternel confond le diagnostic néonatal du syndrome de Timothée de type 1. Pédiatrie (2013) 131: e1991–5. doi: 10.1542/ peds.2012-2941
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
70. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. La cavéoline-3 mutante induit un courant de sodium tardif persistant et est associée au syndrome du QT long. Circulation (2006) 114:2104–12. doi: 10.1161 / CIRCULATIONAHA.106.635268
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
71. Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, et al. Nouveau mécanisme pour le syndrome de mort subite du nourrisson: courant de sodium tardif persistant secondaire à des mutations de la cavéoline-3. Rythme cardiaque (2007) 4:161-6. doi: 10.1016/j. hrthm.2006.11.030
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
72. Il s’agit de l’un des plus grands groupes de la planète. Génétique moléculaire de la famille des gènes de la cavéoline: implications pour les cancers humains, le diabète, la maladie d’Alzheimer et la dystrophie musculaire. Am J Hum Genet (1998) 63:1578-87. doi:10.1086/302172
CrossRef Texte Intégral
73. Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. La localisation des canaux Ca (2 +) cardiaques de type L vers un complexe de signalisation macromoléculaire cavéolaire est nécessaire pour la régulation bêta (2)-adrénergique. Proc Natl Acad Sci U S A (2006) 103:7500-5. doi: 10.1073/pnas.0503465103
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
74. Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, et al. Sous-unité beta4 du canal sodique codé SCN4B dans le syndrome congénital du QT long. Circulation (2007) 116:134-42. doi: 10.1161 / CIRCULATIONAHA.106.659086
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
75. Chen L, Marquardt ML, Testeur DJ, Sampson KJ, Ackerman MJ, Kass RS. La mutation d’une protéine d’ancrage à l’A-kinase provoque un syndrome du QT long. Proc Natl Acad Sci U S A (2007) 104:20990-5. doi: 10.1073/pnas.0710527105
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
76. Wu G, Ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, et al. Mutation de l’alpha-1-syntrophine et syndrome du QT long: une maladie de perturbation des canaux sodiques. Circ Arythm Electrophysiol (2008) 1:193-201. doi: 10.1161/ CIRCEP.108.769224
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, et al. Identification d’un Kir3.4 mutation dans le syndrome congénital du QT long. Am J Hum Genet (2010) 86:872-80. doi: 10.1016/j.ajhg.2010.04.017
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
78. Roden DM. Mécanismes et gestion de la proarythmie. Am J Cardiol (1998) 82:49I-57I.doi:10.1016/S0002-9149 (98) 00472-X
CrossRef Texte intégral
79. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. Une base structurelle pour le syndrome du QT long induit par le médicament. Proc Natl Acad Sci U S A (2000) 97:12329-33. doi: 10.1073/pnas.210244497
CrossRef Texte intégral
80. Kannankeril PJ, Roden DM. QT long induit par le médicament et torsade de pointes: progrès récents. Curr Opin Cardiol (2007) 22:39-43. doi: 10.1097/HCO.0b013e32801129eb
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
81. Veerman CC, Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken CA, et al. Le blocage lent du courant potassique du redresseur retardé contribue de manière importante au syndrome du QT long induit par le médicament. Circ Arythm Electrophysiol (2013) 6:1002-9. doi: 10.1161/ CIRCEP.113.000239
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
82. Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. Un polymorphisme commun associé à une arythmie cardiaque induite par des antibiotiques. Proc Natl Acad Sci U S A (2000) 97:10613-8. doi: 10.1073/pnas.180223197
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
83. Kääb S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, et al. Une vaste étude de gènes candidats identifie le polymorphisme KCNE1 D85N comme un modulateur possible des torsades de pointes induites par le médicament. Circ Cardiovasc Genet (2012) 5:91-9. doi: 10.1161/ CIRCGÉNÉTIQUE.111.960930
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
84. Nakamura K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase S, et al. Syndrome du QT long induit par les anticorps Anti-KCNH2: nouvelle forme acquise du syndrome du QT long. J Am Coll Cardiol (2007) 50:1808-9. doi: 10.1016 / j.jacc.2007.07.037
CrossRef Texte intégral
85. Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, et al. Modèles d’ondes T ECG dans des formes génétiquement distinctes du syndrome héréditaire du QT long. Circulation (1995) 92:2929-34. doi: 10.1161/01.CIR.92.10.2929
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
86. Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giulia LC, et al. Spectre des modèles d’ondes ST-T et des paramètres de repolarisation dans le syndrome congénital du QT long: Les résultats de l’ECG identifient les génotypes. Circulation (2000) 102:2849-55. doi: 10.1161/01.CIR.102.23.2849
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
87. Tan HL, Bardai A, Shimizu W, Moss AJ, Schulze-Bahr E, Noda T, et al. Apparition d’arythmies spécifiques au génotype dans le syndrome congénital du QT long: implications thérapeutiques possibles. Circulation (2006) 114:2096-103. doi: 10.1161 / CIRCULATIONAHA.106.642694
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
88. Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Différences liées à l’âge et au sexe dans les manifestations cliniques chez les patients atteints du syndrome congénital du QT long: résultats du Registre international du LQTS. Circulation (1998) 97:2237-44. doi: 10.1161/01.CIR.97.22.2237
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
89. Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, et al. Influence du génotype sur l’évolution clinique du syndrome du QT long. Groupe de Recherche International Sur Le Registre du Syndrome du QT Long. (1998) 339:960-5. doi: 10.1056 / NEJM199810013391404
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
90. Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson DR, McNitt S, et al. Syndrome du QT long après l’âge de 40 ans. Circulation (2008) 117: 2192-201. doi: 10.1161 / CIRCULATIONAHA.107.729368
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
91. Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, et al. Registre du syndrome. Moduler les effets de l’âge et du sexe sur l’évolution clinique du syndrome du QT long par génotype. J Am Coll Cardiol (2003) 42:103-9. doi: 10.1016/S0735-1097(03)00554-0
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
92. Seth R, Moss AJ, McNitt S, Zareba W, Andrews ML, Qi M, et al. Syndrome du QT long et grossesse. J Am Coll Cardiol (2007) 49:1092-8. doi: 10.1016 / j.jacc.2006.09.054
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
93. Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, et al. Un lien moléculaire entre le syndrome de mort subite du nourrisson et le syndrome du QT long. (2000) 343:262-7. doi: 10.1056/NEJM200007273430405
CrossRef Texte intégral
94. Schwartz PJ, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, et al. Diagnostic moléculaire chez un enfant atteint du syndrome de mort subite du nourrisson. Lancette (2001) 358:1342-3. doi: 10.1016/S0140-6736(01)06450-9
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
95. Christiansen M, Tønder N, Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutations dans le canal ionique K + de HERG: un nouveau lien entre le syndrome du QT long et le syndrome de mort subite du nourrisson. Am J Cardiol (2005) 95:433-4. doi: 10.1016/j. carte amj.2004.09.054
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
96. DJ testeur, MJ Ackerman. Syndrome post-mortem du QT long test génétique pour la mort subite inexpliquée chez les jeunes. J Am Coll Cardiol (2007) 49:240-6. doi: 10.1016 / j.jacc.2006.10.010
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
97. Hofman N, Tan HL, Clur SA, Alders M, van Langen IM, Wilde AA. Contribution de la maladie cardiaque héréditaire à la mort cardiaque subite dans l’enfance. Pediatrics (2007) 120: e967–73. doi: 10.1542/ peds.2006-3751
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
98. W. W., M. J. Smits, M. Schulze-Bahr, M. Arnold, M. Veldkamp, M. Bajanowski, et coll. Mutation de novo dans le gène SCN5A associée à l’apparition précoce de la mort subite du nourrisson. Circulation (2001) 104:1158-64. doi: 10.1161/hc3501.095361
CrossRef Texte intégral
99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, et al. Dysfonctionnement cardiaque des canaux sodiques dans le syndrome de mort subite du nourrisson. Circulation (2007) 115:368-76. doi: 10.1161 / CIRCULATIONAHA.106.646513
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
100. Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Caractérisation moléculaire et fonctionnelle de nouvelles mutations du gène de type glycérol-3-phosphate déshydrogénase 1 (GPD1-L) dans le syndrome de mort subite du nourrisson. Circulation (2007) 116:2253-9. doi: 10.1161 / CIRCULATIONAHA.107.704627
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
101. Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, et al. Mutations associées au syndrome de mort subite du nourrisson dans les sous-unités bêta du canal sodique. Rythme cardiaque (2010) 7:771-8. doi: 10.1016/j. hrthm.2010.01.032
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
102. Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, et al. Perte fœtale récurrente au troisième trimestre et mosaïcisme maternel pour le syndrome du QT long. Circulation (2004) 109:3029-34. doi: 10.1161/01.CIR.0000130666.81539.9E
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
103. Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Tous les bêta-bloquants ne sont pas égaux dans la prise en charge du syndrome du QT long de types 1 et 2: récurrences plus élevées d’événements sous Métoprolol. Je suis Coll Cardiol (2012) 60:2092-6. doi: 10.1016 / j.jacc.2012.07.046
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
104. Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Dénervation sympathique cardiaque gauche dans la prise en charge des patients à haut risque atteints du syndrome du QT long. Circulation (2004) 109: 1826-33. doi: 10.1161/01.CIR.0000125523.14403.1E
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
105. Singh B, al Shahwan SA, Habbab MA, al Deeb SM, Biary N. Syndrome idiopathique du QT long: poser la bonne question. Lancette (1993) 341:741. doi:10.1016/0140-6736(93)90501-7
CrossRef Texte Intégral
106. Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. Le syndrome du QT long avec conduction auriculo-ventriculaire altérée: une variante maligne chez les nourrissons. J Cardiovasc Electrophysiol (1998) 9:1225-32. doi: 10.1111/j.1540-8167.1998.tb00096.x
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
107. Kantoch MJ, Qurashi MM, Bulbul ZR, Gorgels AP. Un nouveau-né avec une cardiopathie congénitale complexe, un bloc auriculo-ventriculaire et une tachycardie ventriculaire torsade de pointes. Pacing Clin Electrophysiol (1998) 21:2664-7. doi: 10.1111/j.1540-8159.1998.tb00043.x
CrossRef Texte intégral
108. El-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Consanguinité parmi la population saoudienne. J Med Genet (1995) 32:623-6. doi: 10.1136/jmg.32.8.623
CrossRef Texte intégral
109. El Mouzan MI, Al Salloum AA, Al Herbish AS, Qurachi MM, Al Omar AA. Consanguinité et troubles génétiques majeurs chez les enfants saoudiens: une étude transversale communautaire. Ann Saudi Med (2008) 28:169-73. doi:10.4103/0256-4947.51726
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
110. Medeiros-Domingo A, Bhuiyan ZA, Testeur DJ, Hofman N, Bikker H, van Tintelen JP, et al. Le récepteur de la ryanodine codé en RYR2 / canal de libération de calcium chez des patients diagnostiqués précédemment avec une tachycardie ventriculaire polymorphe catécholaminergique ou un syndrome du QT long induit par l’exercice induit par un génotype négatif: une analyse mutationnelle complète du cadre de lecture ouvert. Je suis Coll Cardiol (2009) 54:2065-74. doi: 10.1016 / j.jacc.2009.08.022
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
111. Al-Aama JY, Al-Ghamdi S, Bdier AY, Wilde AA, Bhuiyan ZA. Mutation de novo dans le gène KCNQ1 causale du syndrome de Jervell et de Lange-Nielsen. Clin Genet (2013). doi: 10.1111/cge.12300
Résumé Pubmed / Texte Intégral Pubmed | Texte Intégral CrossRef
112. Il s’agit de l’un des plus grands noms de la littérature française. Variante du canal sodique SCN5A impliquée dans le risque d’arythmie cardiaque. Science (2002) 297:1333-6. doi: 10.1126 / science.1073569
CrossRef Texte intégral
113. Wilde AA, Bezzina CR. Génétique des arythmies cardiaques. Cœur (2005) 91:1352-8.