Gestion de l’hyponatrémie chez les patients atteints d’insuffisance cardiaque
L’utilisation de Vaptans dans le traitement de l’Hyponatrémie
Les actions AVP sont médiées par une interaction du peptide avec une série de récepteurs situés sur des cellules dans tout le corps. Les vaptans sont des agents non peptidergiques qui bloquent l’interaction de l’AVP avec ces récepteurs; ils sont classés selon le sous-type de récepteur qu’ils affectent. Comme mentionné précédemment, l’activation du récepteur V2 sur les cellules tubulaires rénales augmente la perméabilité du canal collecteur à l’eau et conduit à la réabsorption de l’eau libre.35 Le récepteur V1A est situé sur les cellules musculaires lisses vasculaires où il médie une augmentation du tonus vasomoteur. Les récepteurs V1A se trouvent également sur les plaquettes et dans le myomètre où ils médient respectivement l’agrégation et la contraction utérine. Certains des antagonistes de l’AVP (par exemple, le conivaptan) bloquent à la fois les récepteurs V1A et V2 tandis que d’autres (par exemple, le tolvaptan et le lixivaptan) sont sélectifs pour le récepteur V2.
Le tolvaptan, un agent sélectif V2, a été largement étudié dans l’insuffisance cardiaque ainsi que chez les patients atteints d’hyponatrémie pour diverses causes. L’une des études initiales a été réalisée chez un groupe de 254 patients insuffisants cardiaques qui ont été randomisés pour recevoir du tolvaptan à des doses allant de 30 mg à 60 mg par jour ou un placebo.36 Le tolvaptan à toutes les doses étudiées a été associé à une réduction significative du poids corporel et à une amélioration des signes et symptômes de l’insuffisance cardiaque. Toutes les doses ont également été associées à une augmentation des taux sériques de sodium dans cette étude. Les patients hyponatrémiques représentaient 28% de la population étudiée et ces patients ont connu la plus forte augmentation du sodium sérique. Il convient de noter que dès le jour 1 de l’étude, 80% des patients atteints de tolvaptan (par opposition à 40% du placebo) avaient une normalisation de leur taux sérique de sodium. Ces effets se sont produits sans changements significatifs de la pression artérielle ou de la fonction rénale et les principaux effets secondaires observés étaient la polyurie, la bouche sèche et la soif. Cette étude a été suivie par l’essai ACTIV in CHF qui comprenait une population légèrement plus importante de 319 patients (dont 21.3% étaient hyponatrémiques à l’inclusion) qui ont été hospitalisés en raison d’une insuffisance cardiaque décompensée.17 Le poids corporel moyen a diminué significativement plus chez les patients traités par le tolvaptan que chez ceux ayant reçu le placebo. Les patients traités par le Tolvaptan ont également connu des augmentations de sodium sérique qui étaient les plus importantes chez les patients hyponatrémiques au début du traitement. Ces changements ont persisté pendant toute la durée de l’étude. Lors de l’analyse post hoc, la survie sans événement avait tendance à être plus longue pour le groupe combiné de patients traités par le tolvaptan que pour le placebo, mais il n’y avait aucune différence dans le taux de réhospitalisation ou de visites imprévues pour insuffisance cardiaque. Comme dans l’étude initiale, le tolvaptan était bien toléré, la bouche sèche étant le principal effet secondaire. Il n’y avait pas d’effets hémodynamiques ou rénaux significatifs.
L’efficacité de l’antagonisme de la vasopressine dans l’étude des résultats de l’insuffisance cardiaque avec l’étude tolvaptan (EVEREST) a randomisé 4133 patients hospitalisés pour insuffisance cardiaque décompensée recevant soit 30 mg de tolvaptan par jour, soit un placebo en plus de leur traitement standard. L’objectif à court terme de l’étude était d’évaluer les effets du traitement sur un point final composite des caractéristiques cliniques globales évaluées par le patient et de la perte de poids au jour 7 (ou au moment de la sortie de l’hôpital) après le début du traitement.37 Les résultats de l’EVEREST ont montré que les patients traités par le tolvaptan présentaient une amélioration plus importante du point final primaire composite. Cet effet a été entraîné par une plus grande réduction du poids corporel avec un médicament actif. Bien que les changements de l’état clinique global ne diffèrent pas entre les groupes d’étude, les patients traités par le tolvaptan ont signalé une amélioration significativement plus importante de la dyspnée au jour 1. Dans 1 (mais pas l’autre des 2 essais de composants de l’EVEREST), il y avait également une amélioration de l’œdème. Au jour 1 et à la sortie, le groupe tolvaptan avec hyponatrémie (défini comme un sodium sérique inférieur à 134 mEq / L) a montré une augmentation significativement plus importante du sodium sérique que chez les patients sous placebo hyponatrémique. Le tolvaptan a été bien toléré et la fréquence des événements indésirables graves était similaire entre les groupes, sans insuffisance rénale excessive ni hypotension.
Les patients inscrits à l’EVEREST ont ensuite été suivis pendant une moyenne de 9.9 mois sous tolvaptan ou placebo afin d’évaluer les effets du traitement sur les deux paramètres primaires de mortalité toutes causes confondues (supériorité et non‐infériorité) et de décès CV ou d’hospitalisation en insuffisance cardiaque.38 Les résultats n’ont montré aucune différence significative dans les résultats de morbidité et de mortalité primaires ou secondaires entre les patients traités par tolvaptan et ceux traités par placebo. Chez les patients de l’EVEREST dont les taux sériques de sodium à l’inclusion étaient inférieurs à 134 mEq /L, il y avait une augmentation significative de 5,49 mEq /L 5,77 mEq / L (SD moyenne) au jour 7 ou à la sortie, si elle était antérieure, avec le tolvaptan, par rapport à 1.85 mEq / L 5,10 mEq /L dans le groupe placebo. Cet effet a été observé dès le jour 1 et s’est maintenu tout au long des 40 semaines de traitement. Les effets secondaires étaient minimes. Dans l’ensemble, le tolvaptan a augmenté la soif et la bouche sèche, mais la fréquence des événements indésirables majeurs était similaire dans les 2 groupes.
Deux essais parallèles multicentriques, randomisés, en double aveugle, contrôlés contre placebo, appelés collectivement l’étude des niveaux ascendants de Tolvaptan dans les hyponatrémies 1 et 2 (SALT‐1 et SALT‐2), ont examiné l’effet du tolvaptan sur l’hyponatrémie hypervolémique et euvolémique de causes diverses.39 Les 448 patients inclus dans les 2 études ont été assignés au hasard à un placebo ou à du tolvaptan à partir d’une dose de 15 mg par jour (augmentant à 30 mg puis à 60 mg si nécessaire, en fonction des concentrations sériques de sodium) et suivis sur une période de 30 jours. La population comprenait 138 patients (31%) présentant une insuffisance cardiaque chronique comme cause d’hyponatrémie, le reste de la population étant divisé entre des patients atteints de cirrhose ou d’un syndrome d’hypersécrétion hormonale antidiurétique inappropriée (SIADH) et d’autres causes d’hyponatrémie. Les 2 principaux critères d’évaluation pour tous les patients étaient la variation de la surface journalière moyenne sous la courbe de la concentration sérique de sodium de l’inclusion au jour 4 et la variation de l’inclusion au jour 30. Comme le montre la figure 3, les concentrations sériques de sodium ont augmenté significativement plus dans le groupe tolvaptan que dans le groupe placebo au cours des 4 premiers jours et après les 30 jours complets de traitement. Une analyse planifiée des essais sur le SEL a montré que la correction de l’hyponatrémie par le tolvaptan était associée à une amélioration significative de l’état mental autodéclaré, en particulier chez les patients présentant une hyponatrémie marquée ou un SIADH. Les améliorations des scores de santé mentale ont été positivement corrélées avec des modifications du sérum dans les groupes tolvaptan et placebo et inversées après l’arrêt du traitement, suggérant que les déficiences de la fonction mentale associées à l’hyponatrémie peuvent être significativement améliorées en augmentant le sérum. Les principaux effets secondaires du tolvaptan comprenaient une soif accrue, une bouche sèche et une miction accrue. Le tolvaptan a été approuvé par la Food and Drug Administration (FDA) des États-Unis pour le traitement de l’hyponatrémie euvolémique et hypervolémique.
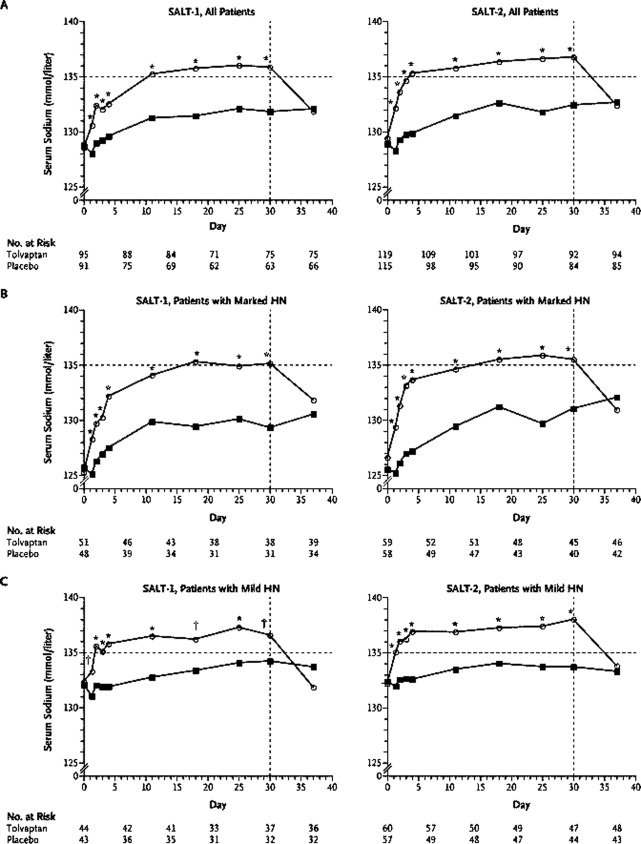 Figure 3
Figure 3
Concentrations sériques moyennes de sodium selon le jour de la visite du patient dans les essais SALT‐1 et SALT‐2. Schrier RW, Gheorghiade M, Berl T, et al. Tolvaptan, un antagoniste sélectif des récepteurs V2 de la vasopressine orale, pour l’hyponatrémie. En anglais J Med. 2006;355:2099–2112. Copyright 2006 Société médicale du Massachusetts. Tous droits réservés. Les astérisques indiquent P < 0,001 pour la comparaison entre le tolvaptan et les patients traités par placebo. Les poignards indiquent P < 0,01 pour la comparaison entre le tolvaptan et le placebo. Le tolvaptan a été arrêté le 30e jour. Les cercles désignent les patients recevant du tolvaptan et les carrés désignent les patients recevant un placebo. Les lignes horizontales indiquent la limite inférieure de la plage normale pour la concentration sérique de sodium. Les lignes verticales indiquent la fin de la période de traitement. HN dénote une hyponatrémie. Abréviation: SEL‐1 / SEL‐2, Étude des Niveaux ascendants de Tolvaptan dans les Hyponatrémies 1 et 2.
Le Lixivaptan est un autre antagoniste sélectif des récepteurs V2 qui a été étudié chez des patients insuffisants cardiaques.40 Dans une étude à dose unique ascendante randomisée, en double aveugle, contrôlée par placebo, 42 patients nécessitant un diurétique présentant une insuffisance cardiaque légère à modérée, les patients ont reçu soit un placebo, soit des doses de lixivaptan allant de 10 mg à 400 mg. À l’exception des patients ayant reçu la dose de 10 mg, le lixivaptan a produit une augmentation significative et liée à la dose du volume urinaire sur une période de 4 heures par rapport au placebo. Sur 24 heures, l’augmentation du volume urinaire était plus importante avec le lixivaptan qu’avec le placebo et ces augmentations s’accompagnaient d’une augmentation significative de l’excrétion d’eau sans soluté. À des doses plus élevées de lixivaptan, les taux sériques de sodium ont augmenté de manière significative. Le médicament était toléré chez ces patients et les effets secondaires avaient tendance à être légers. Le traitement de l’Hyponatrémie à base de Lixivaptan dans l’évaluation des patients cardiaques de classe III / IV de la NYHA (Étude sur L’ÉQUILIBRE) est un essai en cours qui est conçu pour évaluer si le lixivaptan est un agent efficace et sûr pour augmenter le sodium sérique chez les patients insuffisants cardiaques qui sont surchargés de volume et qui souffrent d’hyponatrémie. Les critères secondaires de l’étude BALANCE incluent la mortalité toutes causes confondues, les effets du CV, l’hospitalisation à haute fréquence et la modification aiguë du poids corporel.
Le conivaptan, antagoniste combiné des récepteurs V1A et V2, a été approuvé par la FDA pour le traitement de l’hyponatrémie euvolémique et hypervolémique. Les effets hémodynamiques aigus ont été étudiés chez 142 patients insuffisants cardiaques de classe III et IV de la NYHA. L’administration de 20 mg ou 40 mg de conivaptan a réduit de manière significative le coin de l’artère pulmonaire et les pressions auriculaires droites pendant l’intervalle de 3 heures à 6 heures après l’administration intraveineuse et a considérablement augmenté la production d’urine de manière dose‐dépendante pendant les 4 premières heures après la dose.41 Dans une autre étude, 170 patients hospitalisés pour insuffisance cardiaque aggravée ont été assignés au hasard à un traitement par conivaptan (dose de charge de 20 mg suivie de 2 perfusions continues successives de 24 heures de 40, 80 ou 120 mg / jour) ou par placebo en plus de leur traitement standard.42 À 24 heures, chaque dose de conivaptan avait augmenté la production d’urine significativement plus que le placebo, la différence étant en moyenne de 1,0 à 1,5 L. L’augmentation moyenne du sodium sérique à 24, 48 et 72 heures était significativement plus élevée dans chacun des groupes de conivaptan par rapport au groupe placebo. À 48 heures, le conivaptan a augmenté le sodium sérique de 2,25 mmol /L à 3,27 mmol / L de plus que le placebo. Le conivaptan a été bien toléré chez ces patients hospitalisés en insuffisance cardiaque. Les réactions au site de perfusion de ce médicament administré par voie intraveineuse étaient l’événement indésirable le plus fréquent et l’administration du médicament n’était pas associée à des modifications cliniquement importantes des signes vitaux, à des troubles électrolytiques ou au rythme cardiaque.
Les effets du conivaptan sur les taux sériques de sodium ont été évalués chez 84 patients hospitalisés présentant une hyponatrémie euvolémique ou hypervolémique définie comme une sodium sérique comprise entre 115 mEq/et 129 mEq/L.43 Ces patients ont reçu soit un placebo par voie intraveineuse, soit du conivaptan administré en dose de charge de 20 mg pendant 30 minutes, suivie d’une perfusion de 96 heures de 40 mg/jour ou de 80 mg/jour. Les résultats représentés à la figure 4 montrent que les deux doses de conivaptan ont été associées à des augmentations très significatives sous la courbe sodium‐temps au cours du traitement de 4 jours. De l’inclusion à la fin du traitement, le sodium sérique a augmenté de 0,8 0,8 mEq / L avec le placebo par rapport à 6,3 0,7 mEq / L et 9,4 0,8 mEq /L avec les doses de 40 mg et 80 mg de conivaptan. Le conivaptan était généralement bien toléré, bien que les réactions au site de perfusion aient conduit au retrait de 1 (3 %) et 4 (15 %) des patients ayant reçu du conivaptan à 40 mg/jour et 80 mg/jour, respectivement.
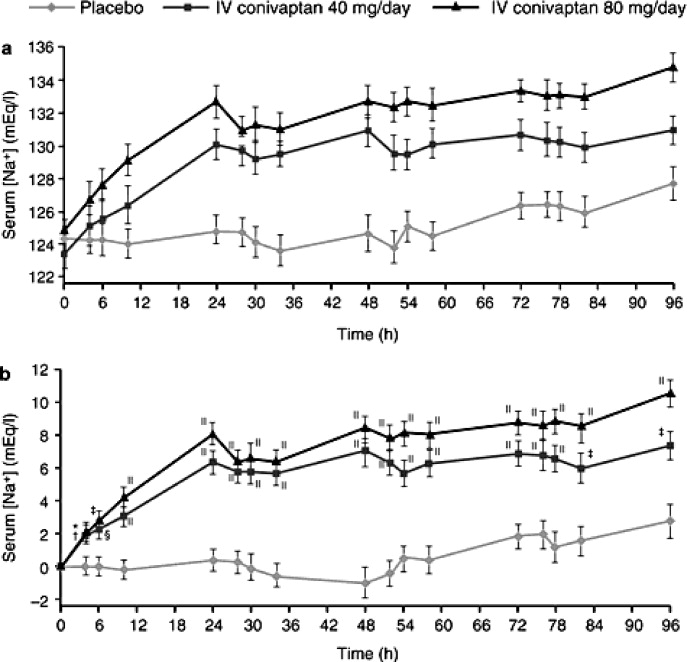 Figure 4
Figure 4
( a) Sérum moyen et (b) changement moyen (LS) par rapport à la valeur initiale dans le sérum à la valeur initiale (heure 0) et à chaque heure de mesure. Les barres T indiquent SE. * P = 0,025; †P = 0.034; ‡P = 0,002; §P = 0,008; ‡P < 0,001.
Le profil de sécurité général des vaptans a été bon. La plupart des effets indésirables, y compris la soif, la bouche sèche et d’autres, ont été mineurs et ces agents, en général, n’ont que des effets minimes sur la pression artérielle et la fonction rénale. De plus, l’innocuité et la tolérabilité à long terme du tolvaptan ont été démontrées dans l’essai EVEREST. Une préoccupation théorique concernant l’utilisation des vaptans pour traiter l’hyponatrémie est que la correction rapide de l’hyponatrémie à un taux > 12 mEq / L sur 24 heures peut provoquer une démyélinisation osmotique des structures cérébrales avec de graves conséquences neurologiques. Il a été conseillé chez les patients sensibles (y compris ceux souffrant de malnutrition sévère, d’alcoolisme ou d’une maladie hépatique avancée) de corriger les taux de sodium à un taux inférieur. Il est également recommandé que les médicaments soient initiés à l’hôpital et que le sodium sérique soit surveillé pendant le traitement.