Guide des tumeurs conjonctivales
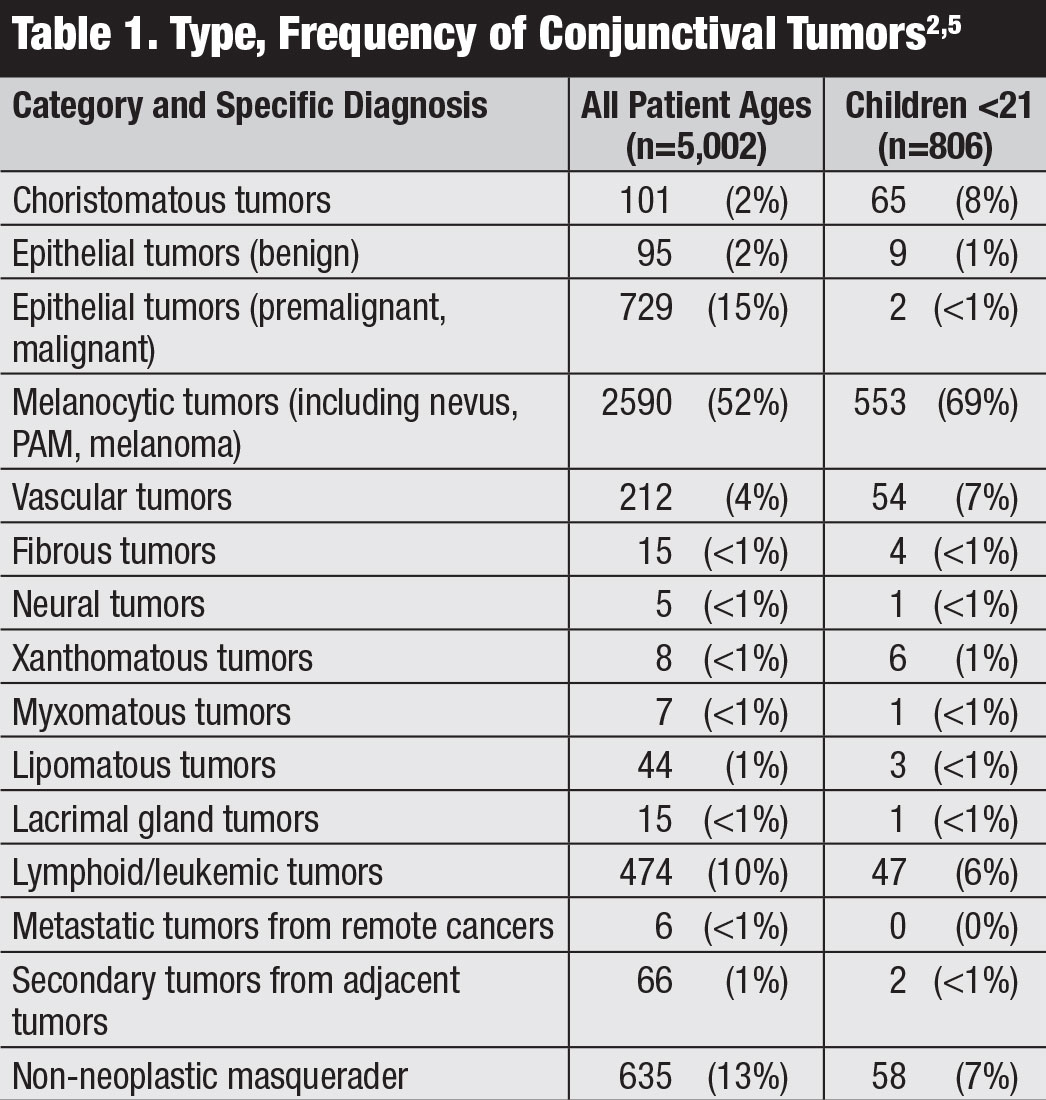
Les tumeurs conjonctivales comprennent un spectre de néoplasmes bénins et malins.1-5 Les types diffèrent en fonction de l’âge et de la race, du statut immunitaire systémique et des expositions à long terme. Une vaste étude portant sur 5 002 cas d’un centre d’oncologie oculaire a révélé que 52% étaient bénins, 18% étaient prémalignés et 30% étaient malins (tableau 1).1,2 Même si ce rapport provenait d’un centre d’oncologie oculaire et que les tumeurs malignes pourraient être surreprésentées, il est important que les cliniciens comprennent la variété des tumeurs conjonctivales.
Les cinq tumeurs les plus courantes étaient le naevus (23%), la néoplasie épidermoïde de la surface oculaire (OSSN, 14%), la mélanose primaire acquise (PAM, 12%), le mélanome (12%) et la tumeur lymphoïde (9%).5 Tumeurs malignes ont été observées le plus souvent chez les adultes et comprenaient un mélanome (12%), un carcinome épidermoïde (CSC, 9%), un lymphome (7%), un sarcome de Kaposi (< 1%), des métastases (< 1%) et d’autres.1 Les tumeurs conjonctivales chez les enfants ne présentent une tumeur maligne que 3% du temps.5
Cet examen des tumeurs conjonctivales les plus courantes vous préparera à les gérer de manière appropriée, que ce soit dans votre bureau ou par aiguillage.
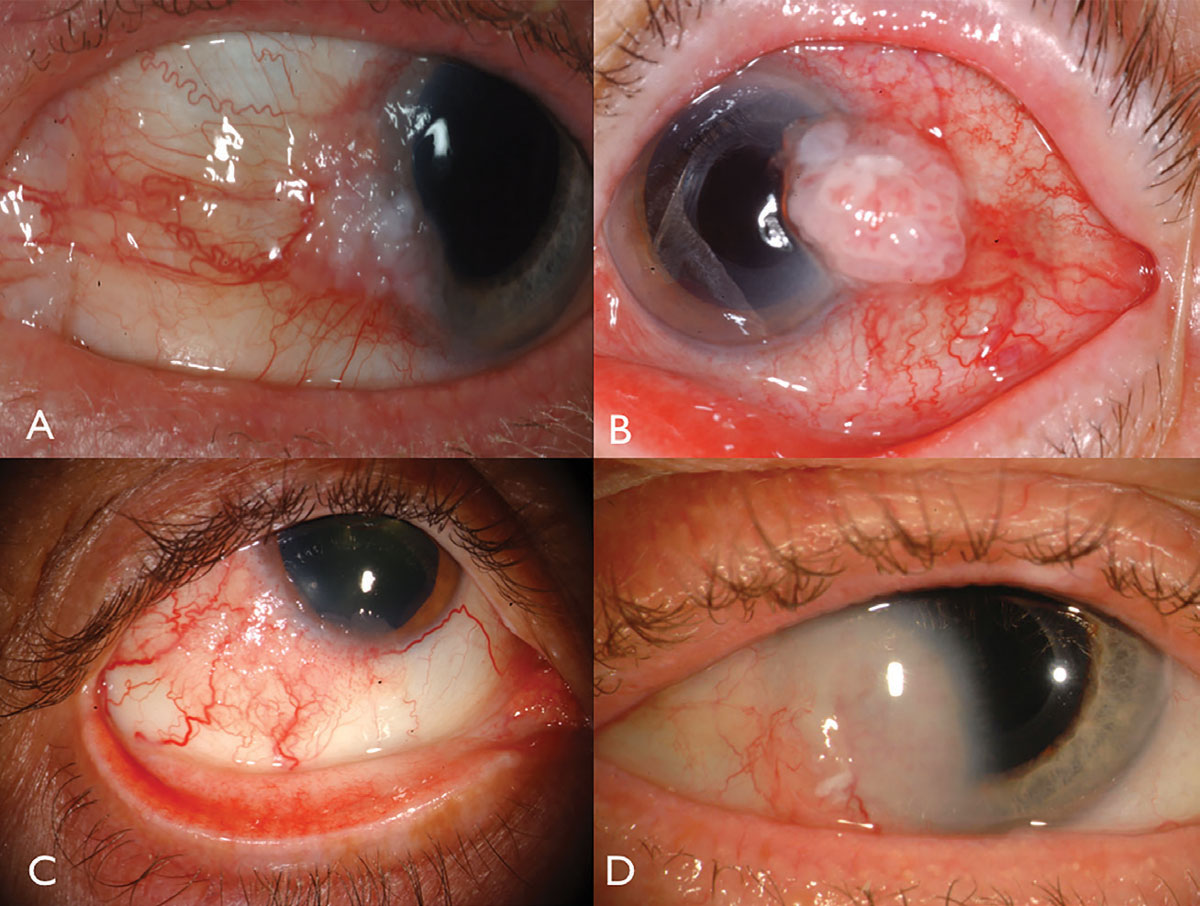
Fig. 1. OSSN LIMBAL: avec leucoplasie et atteinte cornéenne (A), avec vascularisation intrinsèque proéminente et vaisseaux nourriciers (B), chez un patient séropositif (C), et avec invasion cornéenne profonde nécessitant une résection et une radiothérapie de la plaque (D). Cliquez sur l’image pour l’agrandir.
Néoplasie épidermoïde de la surface oculaire
Le terme clinique général d’OSSN comprend un spectre de tumeurs malignes allant de modifications dysplasiques épithéliales légères, telles que la néoplasie intra-épithéliale conjonctivale (NIC), à un carcinome invasif plus grave qui envahit la membrane basale dans la substantia propria, tel que le carcinome épidermoïde.
Caractéristiques cliniques. L’OSSN conjonctival se produit classiquement chez les hommes caucasiens plus âgés, en particulier ceux qui sont exposés au soleil de manière chronique. Aux États-Unis, le CSC conjonctival est cinq fois plus fréquent chez les hommes et les Caucasiens. Cependant, en Afrique, le CSC conjonctival est presque aussi fréquent chez les hommes et les femmes, et il survient à un âge plus jeune qu’aux États-Unis.6
La néoplasie épidermoïde de la surface oculaire se présente généralement sous la forme d’une masse gélatineuse vascularisée unilatérale, située dans la conjonctive exposée au soleil au niveau du limbe nasal ou temporal (Figure 1). Une leucoplasie sus-jacente, des vaisseaux d’alimentation dilatés et une infiltration mousseuse de l’épithélium cornéen adjacent peuvent se produire et peuvent rarement envahir le globe ou l’orbite.
Cliquez sur tableau pour l’agrandir.
Facteurs prédisposants. Les facteurs environnementaux les plus importants pour l’OSSN comprennent l’exposition chronique au soleil et l’exposition à la fumée de cigarette. Deux facteurs clés prédisposant l’hôte comprennent le teint clair et le virus de l’immunodéficience humaine (VIH) sous-jacent et le virus du papillome humain.6 Les patients immunodéprimés, en particulier ceux atteints du VIH, sont à risque d’être atteints d’OSSN et peuvent présenter des tumeurs avancées, bilatérales et invasives.1 C’est particulièrement le cas en Afrique, où le VIH est répandu et où l’OSSN se produit aussi bien chez les hommes que chez les femmes et à un plus jeune âge.6 D’autres dérèglements immunitaires peuvent prédisposer un patient à l’OSSN, y compris l’immunosuppression par greffe d’organe, l’eczéma / atopie, la pemphigoïde cicatricielle oculaire, la xéroderma pigmentosum et des affections auto-immunes.7
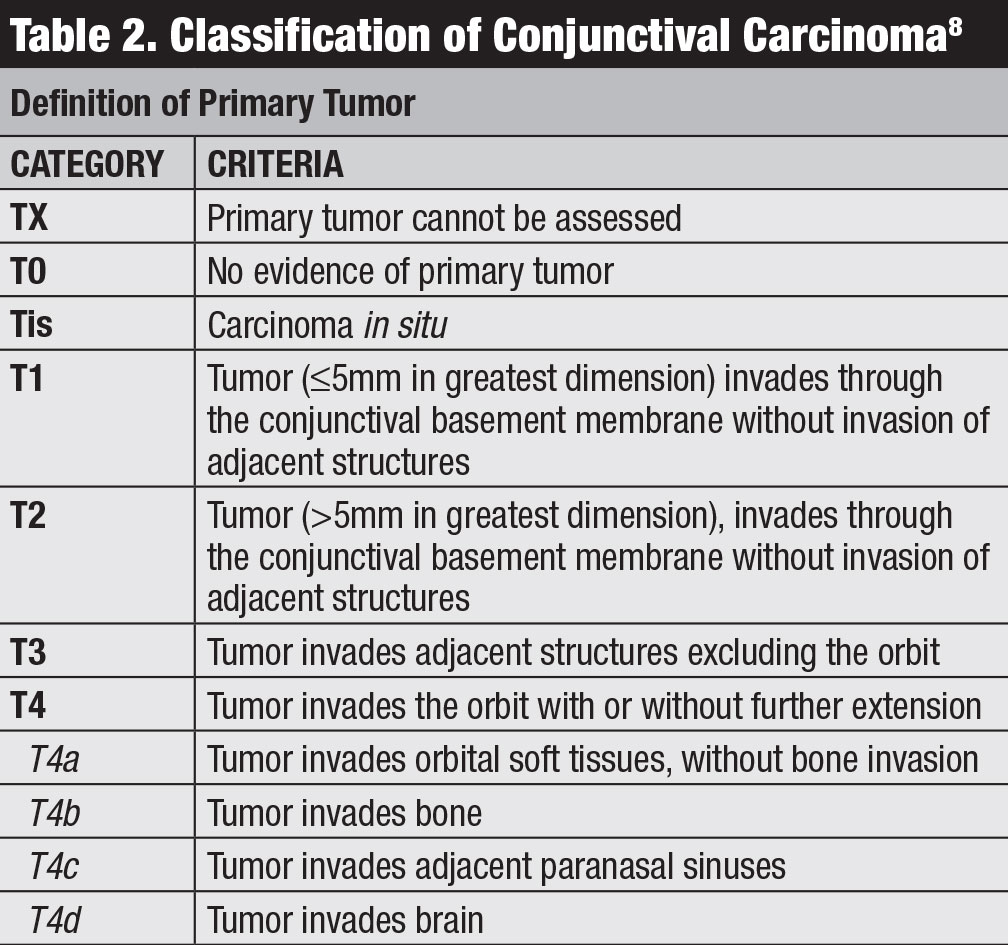
Classification. La 8e édition du manuel de l’American Joint Committee on Cancer (AJCC) fournit la classification la plus récente du carcinome conjonctival, y compris le CSC et le CIN (tableau 2).8
Gestion. Cela implique une résection chirurgicale utilisant la technique “sans contact” ou des thérapies non chirurgicales telles que la chimiothérapie topique avec la mitomycine C (MMC) ou le 5-fluorouracile (5-FU), une immunothérapie topique ou injectée avec l’interféron alpha-2b (IFN), un médicament antiviral topique (cidofovir) ou une thérapie photodynamique.7,9-11
La technique chirurgicale sans contact implique une évaluation détaillée de la tumeur à l’aide d’une biomicroscopie à lampe à fente pour visualiser toutes les marges tumorales, y compris les composants bulbaires, fornicaux et tarsiens, pour comprendre toute l’étendue de la tumeur et permettre au clinicien de dessiner à la main un enregistrement de modèle.9 Ce modèle est ensuite pris dans la chirurgie pour s’assurer que la tumeur entière est retirée.
Au moment de la chirurgie, seul le tissu normal environnant est maintenu avec une pince et la tumeur n’est jamais touchée pour éviter l’ensemencement de la tumeur. De plus, une solution saline équilibrée n’est pas utilisée pendant la chirurgie pour éviter la dispersion liquide des cellules cancéreuses. Après l’ablation de la tumeur, la fermeture avec des instruments propres est cruciale. En utilisant cette technique pour l’OSSN, la persistance ou la récurrence tumorale se retrouve dans moins de 5% des cas.
Cliquez sur tableau pour l’agrandir.
La chimiothérapie topique avec 5-FU ou MMC est efficace pour résoudre l’OSSN, souvent dans les deux à quatre semaines suivant le traitement, bien qu’un risque de carence en cellules souches existe. Notre préférence thérapeutique topique est l’immunothérapie par IFN, car elle est bien tolérée avec un bon contrôle tumoral, souvent sur trois mois et avec peu de complications et seulement une conjonctivite folliculaire mineure.10,11 Ces médicaments peuvent être localement toxiques pour l’épithélium cornéen, mais moins avec l’interféron, et les patients doivent être suivis de près pendant leur traitement. Si le coût est un facteur pour le patient, le 5-FU topique est le moins cher, suivi du MMC puis de l’IFN.
Tumeurs lymphoïdes conjonctivales
Les néoplasmes lymphoïdes vont de tumeurs de bas à haut grade et résultent de la prolifération monoclonale des lymphocytes. Les tumeurs lymphoïdes qui se produisent dans la région périoculaire impliquent souvent plusieurs tissus tels que la conjonctive, l’orbite et la paupière et sont appelées tumeurs lymphoïdes “annexes oculaires”, y compris l’hyperplasie lymphoïde réactive bénigne (BRLH) et le lymphome.
La BRLH et le lymphome se trouvent aux extrémités opposées du spectre, la BRLH apparaissant cliniquement comme une “tache de saumon” localisée et histopathologiquement bénigne, tandis que le lymphome apparaît également comme une “tache de saumon” mais avec des caractéristiques histopathologiques plus agressives, avec une activité mitotique et classée comme maligne.
Les tumeurs lymphoïdes annexes oculaires sont généralement d’origine à cellules B. Une étude multicentrique de 268 patients atteints de lymphome conjonctival a révélé que les quatre types les plus courants comprenaient le lymphome de la zone marginale extranodale (ENMZL, précédemment appelé tissu lymphoïde associé à la muqueuse) chez 68%, le lymphome folliculaire (FL) chez 16%, le lymphome à cellules du manteau (MCL) chez 7% et le lymphome diffus à grandes cellules B (DLBCL) chez 5%.12 Les autres types de lymphome conjonctival comprennent le lymphome lymphoplasmocytaire et le plasmocytome.
Caractéristiques cliniques. Le lymphome conjonctival se présente généralement chez les patients âgés de 60 à 70 ans. Cette tumeur peut se manifester par un lymphome primaire, limité à la région périoculaire, ou par un lymphome secondaire avec maladie ailleurs. La plupart des lymphomes primaires se produisent avec ENMZL et FL et les lymphomes secondaires avec DLBCL et MCL. Une analyse de 117 patients atteints d’un lymphome conjonctival a révélé une atteinte systémique chez 31%, le plus souvent chez ceux atteints d’un lymphome oculaire annexal multifocal bilatéral.13
Le lymphome conjonctival se manifeste classiquement sous la forme d’une masse sous-conjonctivale rose de couleur saumon à surface lisse, parfois avec des vaisseaux d’alimentation (figure 2). Cette masse lisse et multilobulée peut ressembler à une conjonctivite folliculaire ou papillaire. Cette tumeur est le plus souvent située dans la région du fornix conjonctival (44%) ou du bulbe médian (42%) et, rarement, dans la caroncule (7%) ou le limbe (7%).13 En plus de la conjonctive, un lymphome peut s’infiltrer dans l’orbite, la paupière ou l’uvée.13 La plupart des patients atteints de lymphome conjonctival ne présentent pas de composant intraoculaire, mais s’il est présent, il réside généralement dans l’uvée et non dans la rétine ou le vitré.
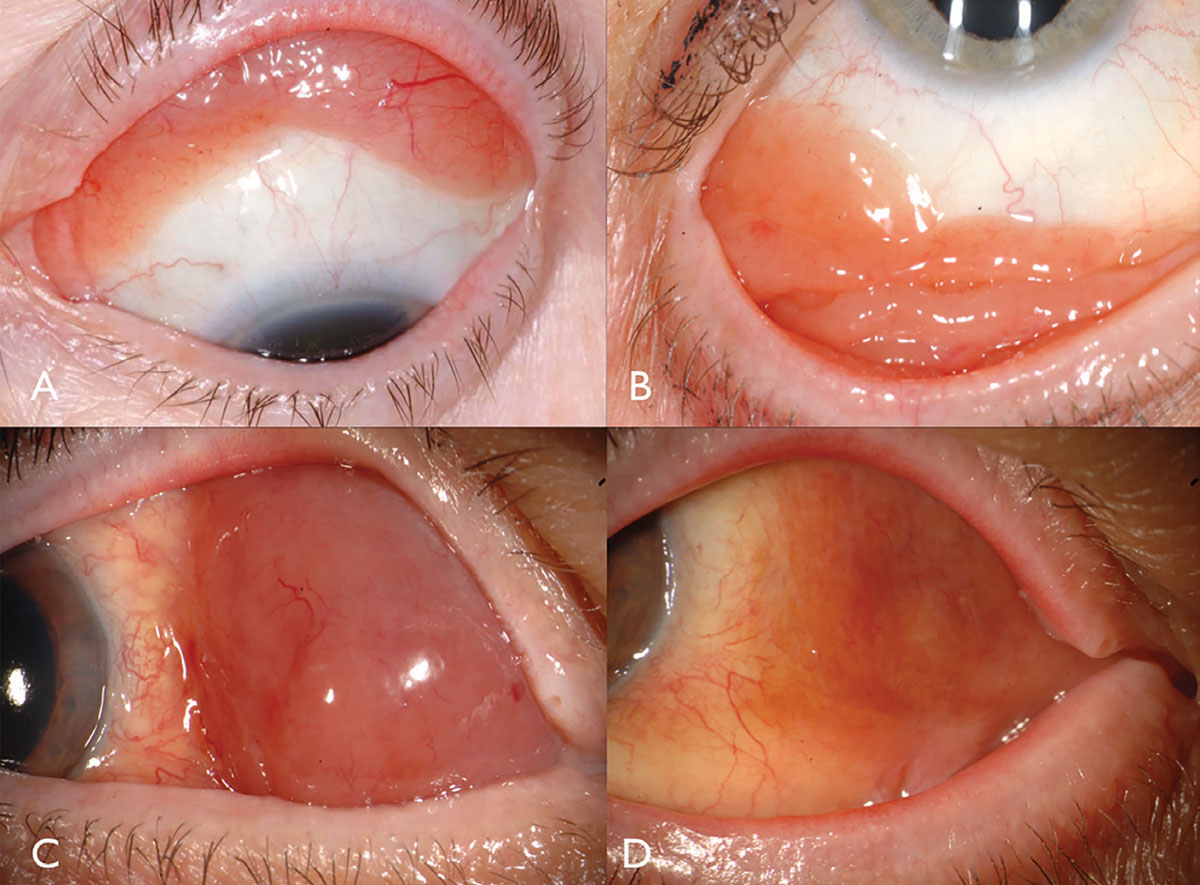
Fig. 2. Conjunctival lymphoma can be salmon-pink (A) or multilobulated forniceal (B). Medial forniceal conjunctival lymphoma before (C) and after (D) ritiximab. Click image to enlarge.
Predisposing factors. Immune dysfunction and autoimmune conditions, as well as infective etiologies such as Helicobacter pylori and Chlamydia psittaci are all predisposing factors for conjunctival lymphoma. La BRLH peut être un précurseur potentiel du lymphome et, bien qu’elle soit principalement présente chez les adultes, elle peut parfois survenir chez les enfants.5 En fait, plus le patient est jeune au moment du diagnostic d’une tumeur lymphoïde conjonctivale, plus il s’agit de BRLH et non d’un lymphome.
Classification. Plusieurs classifications pour le lymphome conjonctival existent, y compris la mise en scène d’Ann Arbor, de l’Organisation mondiale de la Santé et de la 8e édition de l’AJCC (Tableau 3).8 La stadification clinique de l’AJCC est basée sur la localisation de la tumeur, le ganglion lymphatique régional et l’implication à distance.8
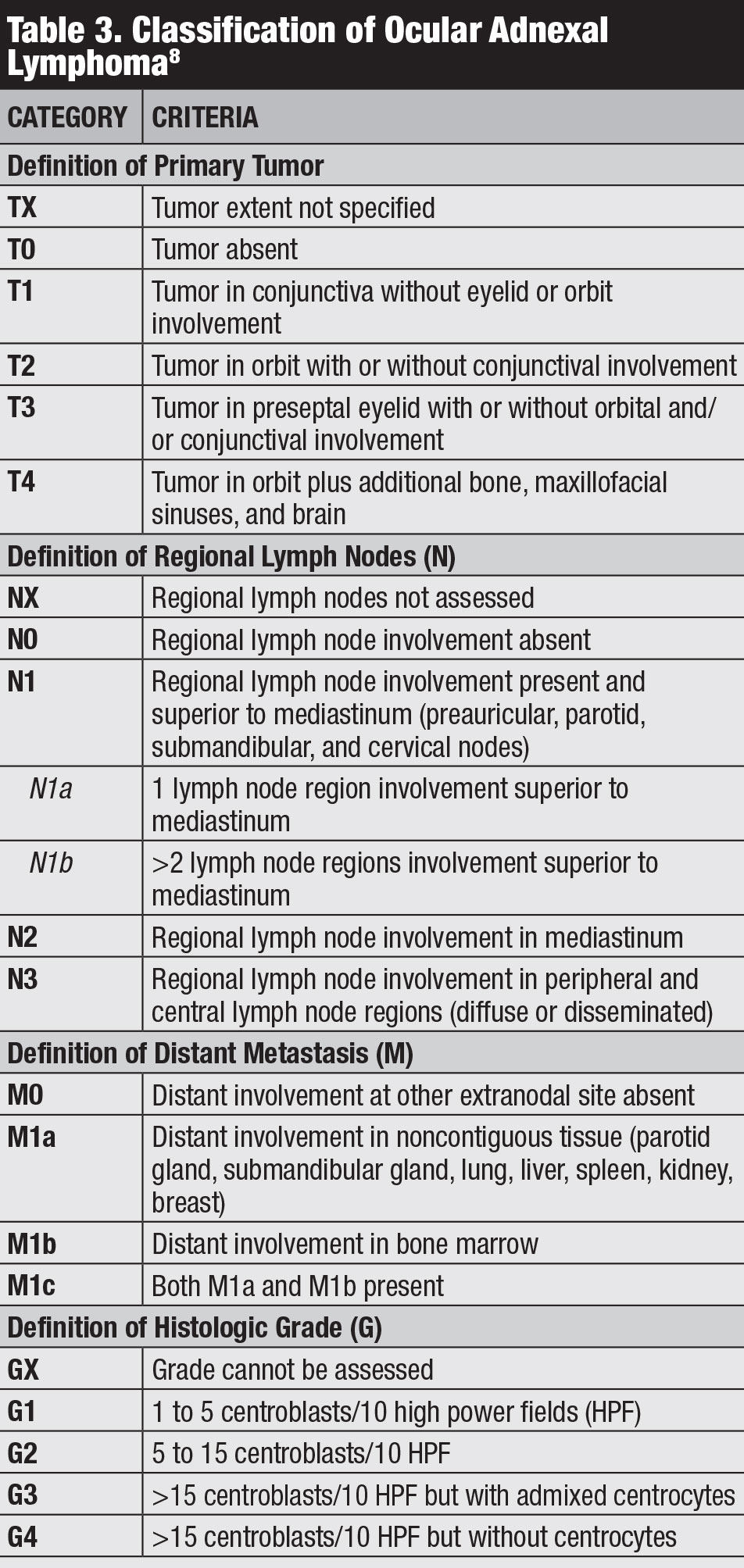
Cliquez sur tableau pour l’agrandir.
Gestion. La prise en charge des patients atteints de lymphome conjonctival dépend principalement de l’étendue de l’atteinte périoculaire, de l’atteinte systémique et de leur état de santé général. Chez les patients atteints uniquement de lymphome conjonctival et sans atteinte systémique, le traitement est axé sur une résection chirurgicale complète. Le traitement par radiothérapie par faisceau externe ou rituximab est une option si la tumeur n’est pas résécable. Pour les personnes atteintes de lymphome périoculaire et systémique, le traitement par rituximab systémique ou l’ajout de chimiothérapie sont des considérations.
Le pronostic systémique avec lymphome conjonctival est directement lié à chaque sous-type, car une étude a révélé que la survie à cinq ans était de 97% pour ENMZL, 82% pour FL, 55% pour DLBCL et seulement 9% pour MCL.12
Mélanome conjonctival
Les tumeurs mélanocytaires conjonctivales sont incontestablement fréquentes, représentant plus de 50% des cas dans une grande série de tumeurs conjonctivales d’une unité d’oncologie oculaire.1,2 Cette classe de tumeurs mélanocytaires comprend de nombreux types tels que naevus, mélanose liée au teint, PAM, mélanose acquise secondaire, mélanome et métastases.1-5 Sur certains continents où les patients ont un teint sombre, même OSSN peut apparaître mélanocytaire. Parmi ces lésions, le naevus conjonctival représente 45% et le mélanome conjonctival primaire représente 23% de toutes les tumeurs mélanocytaires dans une pratique d’oncologie oculaire.2
Aux États-Unis, l’incidence ajustée en fonction de l’âge du mélanome conjonctival a doublé entre 1973 et 1999, passant de 0,27 par million à 0,54 par million.14,15 L’incidence a augmenté de 295% chez les hommes blancs aux États-Unis au cours de la même période de 27 ans, en particulier chez les hommes âgés de 60 ans ou plus.14 Les chercheurs spéculent que le taux croissant peut être lié à l’exposition à la lumière ultraviolette.
Caractéristiques cliniques. Le mélanome conjonctival est une tumeur maligne pigmentée ou non pigmentée pouvant résulter de PAM, naevus ou de novo.16 Le mélanome se trouve sur la conjonctive limbale, bulbaire, fornicéale ou palpébrale et présente souvent des vaisseaux d’alimentation et intrinsèques dilatés et tortueux généralement entourés de PAM plats (figure 3). En général, les tumeurs mesurant plus de 2 mm d’épaisseur présentent un risque important de métastases ganglionnaires. L’invasion tumorale dans l’orbite est particulièrement grave avec un risque métastatique important.
Une récidive tumorale locale ou une nouvelle tumeur se retrouve dans 50% des cas, souvent liée à une nouvelle transformation de PAM. Des métastases à distance – souvent à la chaîne ganglionnaire préauriculaire, sous-maxillaire ou cervicale – sont observées chez 25% des patients. La biopsie ganglionnaire sentinelle peut aider les cliniciens à évaluer l’infiltration ganglionnaire subclinique. Les récurrences multiples, en particulier celles impliquant l’orbite, nécessitent une exentération orbitale.
Facteurs prédisposants. Le facteur prédisposant le plus important pour le mélanome conjonctival est la présence de naevus conjonctival de longue date ou de PAM.16-18 Lors de l’étude de l’origine du mélanome conjonctival par histopathologie, les chercheurs ont découvert que l’origine était PAM dans 74%, de novo dans 19% et naevus dans 7%.16 études cliniques estiment qu’un naevus sur 300 se transforme en mélanome.17,18
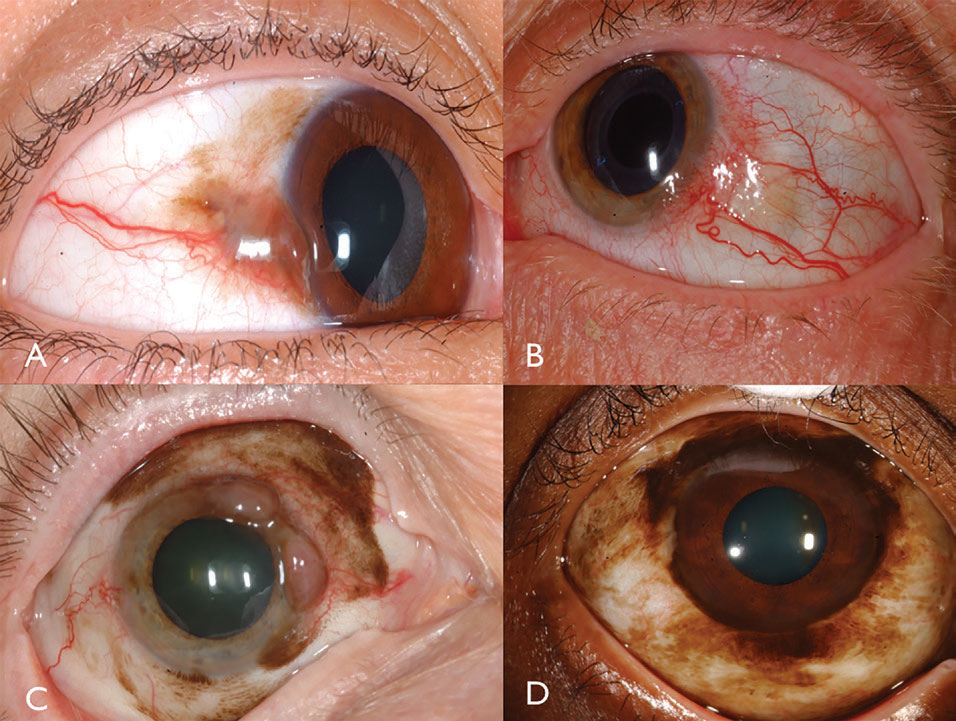
Fig. 3. Le mélanome conjonctival pigmenté peut provenir de PAM (A). Le mélanome conjonctival non pigmenté peut présenter une vascularisation intense (B). La PAM pourrait également provoquer un mélanome conjonctival mixte pigmenté / non pigmenté (C). PAM a provoqué un mélanome limbal chez ce patient afro-américain (D). Cliquez sur l’image pour l’agrandir.
Une vaste étude clinique a révélé que le risque de transformation de la PAM en mélanome sur 10 ans était d’environ 9% et que la plus grande étendue de la PAM favorisait un plus grand risque de transformation en mélanome.19 Par conséquent, il est important d’identifier PAM et de traiter cette affection par une excision chirurgicale, une cryothérapie et même une kératectomie superficielle (s’il y a atteinte cornéenne) dans le but de prévenir le mélanome.
La différenciation du naevus conjonctival du mélanome peut être difficile. Dans une analyse récente de 510 cas de naevus conjonctival vs. mélanome chez les enfants, le mélanome était plus fréquent chez les enfants plus âgés, avec un risque relatif (RR) de 4,80, une plus grande épaisseur tumorale (RR de 1,14), une plus grande base (RR de 4,92), une hémorragie tumorale (RR de 25,30) et des kystes intrinsèques manquants (RR de 5,06).5 Les chercheurs ont attribué ces caractéristiques, prédictives du mélanome conjonctival chez l’enfant, à un mnémonique: ATTRAPER le Mélanome, représentant: Les enfants Vieillissent plus, l’épaisseur / la base plus grande, le kyste manquant, l’hémorragie pour le Mélanome.5
La différenciation de la PAM par rapport au mélanome peut également être difficile; cependant, le mélanome a une épaisseur et la PAM est complètement plate. Dans une analyse de 1 224 cas de PAM par rapport au mélanome chez tous les âges, le mélanome est significativement plus élevé en fonction de l’âge médian du patient (54 par rapport à 61 ans); du sexe masculin (35% par rapport à 49%); de la localisation dans fornix (2% par rapport à 6%) et tarse (1% par rapport à 4%); diamètre basal médian plus grand (6 mm par rapport à 8 mm), épaisseur (< 1 mm par rapport à 1 mm), vaisseaux d’alimentation (10 % contre 48%) et les vaisseaux intrinsèques (4% contre 33%); et hémorragie (< 1% contre 3%).2
Les biomarqueurs tissulaires sont importants pour l’évaluation du mélanome conjonctival et comprennent la mutation BRAF, la mutation du promoteur TERT et la mutation PTEN.1 L’identification de ces biomarqueurs est essentielle lors de la planification d’un traitement systémique pour le traitement ou la prévention des métastases, car des thérapies ciblées contre certains biomarqueurs sont disponibles, comme le vémurafénib pour la malignité mutée par le BRAF.
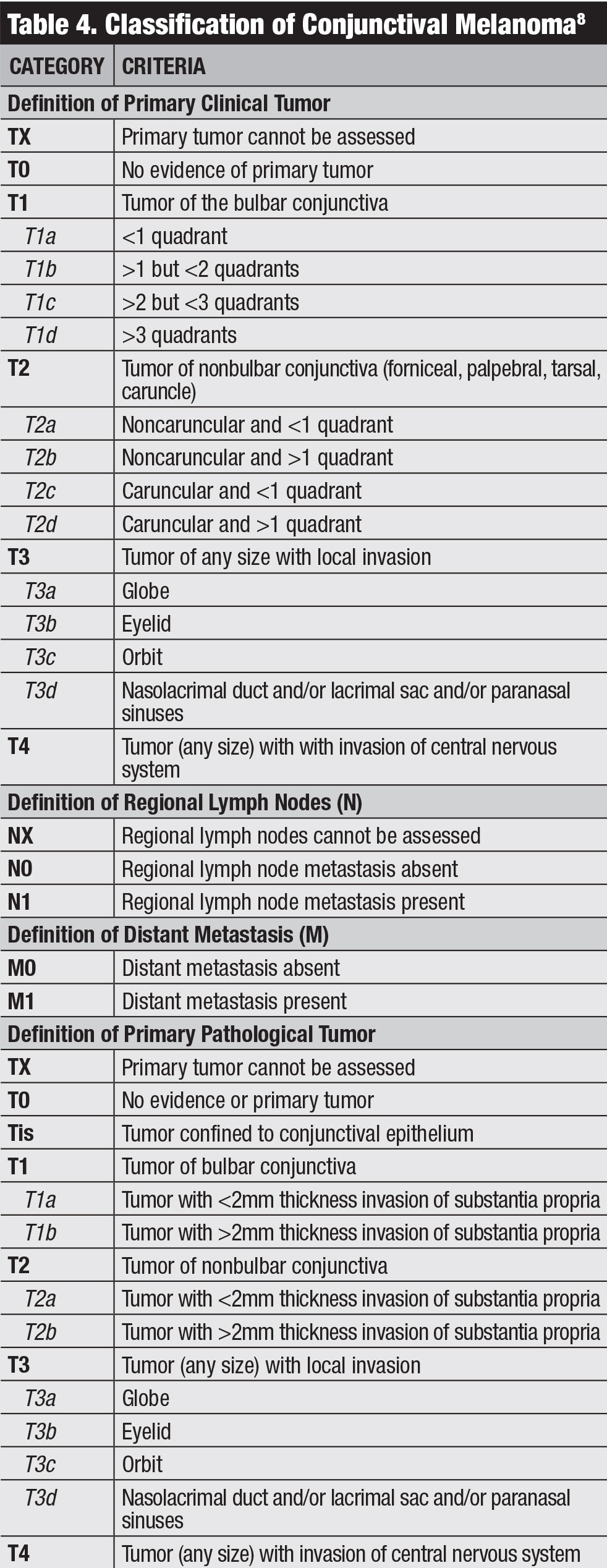
Cliquez sur tableau pour l’agrandir.
Classification. La classification clinique de l’AJCC pour le mélanome conjonctival est basée sur l’étendue de la tumeur par quadrants, la localisation de la tumeur et les caractéristiques invasives (tableau 4).8 Notre équipe a étudié les résultats du mélanome conjonctival sur la base de la 7e édition de l’AJCC et a constaté que cette stadification était hautement prédictive du pronostic.20 Les mélanomes classés comme T2 et T3 (par rapport à T1) ont montré des taux significativement plus élevés de récidive locale, de métastases ganglionnaires régionales, de métastases à distance et de décès.
Gestion. Le traitement du mélanome conjonctival implique essentiellement une résection chirurgicale complète utilisant la technique sans contact pour éviter l’ensemencement tumoral. La première chirurgie est la plus importante, car l’ablation délicate de la tumeur entière sans ensemencement tumoral est essentielle pour prévenir les récidives et les métastases futures.16
Le mélanome au limbe cornéoscléral est enlevé au microscope opératoire en utilisant également la technique sans contact. Le composant cornéen plat est éliminé avec de l’alcool absolu, une épithéliectomie superficielle sans perturbation de la membrane de Bowman. La partie conjonctivale est enlevée avec des marges de 2 à 3 mm et libérée au niveau du limbe à l’aide d’une dissection épisclérale plate. En cas d’invasion sclérale, une radiothérapie par plaque est appliquée. Toutes les marges conjonctivales sont traitées par cryothérapie double gel-dégel.
La reconstruction implique des techniques de fermeture primaires, une greffe de lambeau rotatif ou de membrane amniotique, souvent avec anneau symblépharon avec drapage de membrane amniotique. Le mélanome qui s’étend dans l’orbite nécessite une exentération orbitale ou, plus récemment, une immunothérapie avec inhibition du point de contrôle.21
Les patients atteints de mélanome conjonctival doivent être surveillés par un oncologue oculaire pour la récidive locale et par un oncologue du mélanome systémique pour la maladie métastatique, en particulier avec palpation régionale des ganglions lymphatiques et biopsie sentinelle des ganglions lymphatiques. Les métastases apparaissent initialement dans les ganglions lymphatiques préauriculaires ou sous-maxillaires, puis plus tard dans les poumons et le cerveau. De nouvelles preuves suggèrent que les métastases de mélanome pourraient être sensibles aux inhibiteurs du BRAF ou aux inhibiteurs des points de contrôle immunitaires.21,22
Les tumeurs conjonctivales englobent un large spectre de tumeurs. Les tumeurs malignes les plus courantes comprennent l’OSSN, le lymphome et le mélanome. La reconnaissance des caractéristiques cliniques classiques, la compréhension des précurseurs et la prise en charge rapide et appropriée de ces tumeurs malignes sont importantes pour obtenir les meilleurs résultats pour les patients.
Les Drs Shields, Lally et Shields travaillent au service d’oncologie oculaire du Wills Eye Hospital, Université Thomas Jefferson, à Philadelphie. Soutien fourni par la Fondation de Recherche sur les tumeurs oculaires, Philadelphie.
1. Shields CL, Chien JL, Surakiatchanukul T, et al. Tumeurs conjonctivales: examen des caractéristiques cliniques, des risques, des biomarqueurs et des résultats. La conférence 2017 de J. Donald M. Gass. Asia Pac J Ophthalmol. 2017;6:109-20.
2. Shields CL, Alset AE, Boal NS et al. Tumeurs conjonctivales dans 5002 cas. Analyse comparative des homologues bénins par rapport aux homologues malins. La conférence 2016 de James D. Allen. Am J Ophthalmol. 2017;173:106-33.
3. Boucliers CL, boucliers JA. Tumeurs de la conjonctive et de la cornée. Surv Ophthalmol. 2004;49:3-24.
4. Grossniklaus HE, Green WR, Luckenbach M, et al. Lésions conjonctivales chez l’adulte. Une revue clinique et histopathologique. Cornée. 1987;6:78-116.
5. Shields CL, Sioufi K, Alset AE, et al. Tumeurs conjonctivales chez les enfants. Caractéristiques différenciant les tumeurs bénignes des tumeurs malignes. JAMA Ophthalmol. 2017;135:215-24.
6. Gichuhi S, Sagoo MS, Weiss HA, et al. Epidémiologie de la néoplasie épidermoïde de la surface oculaire en Afrique. Trop Med Int Santé. 2013;18:1424-43.
7. Boucliers CL, Ramasubramanien A, Mellen P, boucliers JA. Carcinome épidermoïde conjonctival survenant chez des patients immunodéprimés (greffe d’organe, infection par le virus de l’immunodéficience humaine). Ophtalmologie. 2011;118:2133-7.
8. Amin MB, Edge S, Greene F, et al., EDS. Manuel de Stadification du cancer AJCC. 8e éd. Il s’agit de la première édition de Springer International Publishing, en 2017.
9. Boucliers JA, Boucliers CL, De Potter PV. Approche chirurgicale des tumeurs conjonctivales. La conférence de Lynn B. McMahan en 1994. Ophthalmol Arch. 1997;115:808-15.
10. Shields CL, Kaliki S, Kim HJ, et al. Interféron pour la néoplasie épidermoïde de la surface oculaire dans 81 cas: Résultats basés sur l’American Joint Committee on Cancer classification. Cornée. 2013;32(3):248-56.
11. Karp CL, Galor A, Chhabra S, et al. Interféron recombinant a2b sous-conjonctival / périlésionnel pour la néoplasie squameuse de la surface oculaire: une revue de 10 ans. Ophtalmologie. 2010;117(12):2241-6.
12. Kirkegaard MM, Rasmussen PK, Coupland SE, et al. Lymphome conjonctival – Une étude rétrospective multicentrique internationale. JAMA Ophthalmol. 2016;134:406-14.
13. Boucliers CL, boucliers JA, Carvalho C, et al. Tumeurs lymphoïdes conjonctivales: analyse clinique de 117 cas et relation avec le lymphome systémique. Ophtalmologie. 2001;108:979-84.
14. Yu GP, Hu DN, McCormick S, Doigt PT. Mélanome conjonctival: Est-ce qu’il augmente aux États-Unis? Am J Ophthalmol. 2003;135:800-6.
15. Tuomaala S, Kivela T. Correspondance concernant le mélanome conjonctival: Augmente-t-il aux États-Unis? Am J Ophthalmol. 2003;136:1189-90.
16. Shields CL, Markowitz JS, Belinsky I, et al. Mélanome conjonctival. Résultats basés sur l’origine tumorale dans 382 cas consécutifs. Ophtalmologie. 2011;118:389-95.
17. Shields CL, Fasiuddin AF, Mashayekhi A, et al. Naevus conjonctivaux: caractéristiques cliniques et évolution naturelle chez 410 patients consécutifs. Ophthalmol Arch. 2004;122:167-75.
18. Gerner N, Norregaard JC, Jensen OA, Prause JU. Naevi conjonctival au Danemark 1960-1980. Une étude de suivi de 21 ans. Acta Ophthalmol Scand. 1996;74:334-7.
19. Shields JA, Shields CL, Mashayekhi A, et al. Mélanose primaire acquise de la conjonctive: risques de progression vers un mélanome dans 311 yeux. La conférence 2006 de Lorenz E. Zimmerman. Ophtalmologie. 2008;115:511-9.
20. Shields CL, Kaliki S, Al-Dahmash S, et al. La classification clinique du Comité mixte américain sur le cancer (AJCC) prédit les résultats du mélanome conjonctival. Ophthalm Plast Reconstr Surg.2012;5:313-23.
21. Sagiv O, Thakar SD, Kandl TJ, et coll. Immunothérapie avec inhibiteurs de la mort cellulaire programmée 1 pour 5 patients atteints de mélanome conjonctival. JAMA Ophthalmol. 1er novembre 2018; 136(11): 1236-41.
22. Dalvin LA, Shields CL, Orloff M, et coll. Immunothérapie inhibitrice des points de contrôle. Indications systémiques et effets secondaires ophtalmiques. Rétine. 2018;6:1063-78.