Ichtyose congénitale autosomique récessive / Actas Dermo-Sifiliográficas
Introduction
La dernière classification consensuelle de l’ichtyose différencie 2 formes principales: les formes non syndromiques, qui se présentent uniquement avec des manifestations cutanées, et les formes syndromiques, qui se présentent également avec des manifestations dans d’autres organes (Tableau 1).1 Parmi les formes non syndromiques, 4 groupes sont identifiés: les ichtyoses communes, les ichtyoses congénitales autosomiques récessives (ARCIs), les ichtyoses kératinopathiques et d’autres ichtyoses moins courantes.Traditionnellement, le groupe des ARCIs était divisé en 2 troubles, l’ichtyose lamellaire (LI) et l’érythrodermie ichtyosiforme congénitale (CIE). Dans la nouvelle classification, l’ichtyose d’arlequin (HI) a été ajoutée à ce groupe1 parce que des mutations inactivantes du gène ABCA12 ont été identifiées comme responsables de ce trouble, 2,3 alors que des mutations non-sens dans le même gène peuvent donner naissance au phénotype LI4 ou CIE5,6. D’autres variantes moins courantes incluses dans le groupe des ARCIs sont le bébé collodion auto-cicatrisant (SHCB), le SHCB acral et l’ichtyose du maillot de bain.7-9
Classification consensuelle Basée sur les caractéristiques cliniques de l’Ichtyose1.
| Formes non dromiques | Formes syndromiques |
| Ichtyose Communechthyose vulgaireéchthyose liée à l’x récessive (non syndromique) Forme ajimajor Ichthyosis de Harlequin Ichthyosisislamellairechthyosis érythrodermaMinor ichthyosiforme congénital Forme de collodion auto-cicatrisant babyAchat de bain collodionacral Ichthyoskératinopathiquechthyosesforme Majeurechthyos épidermolytique Supérieurechthyos épidermolytique supérieurminor formes ichtyose épidermolytique annulaireéchthyose de Macklin-urthyose autosomale récessive ichtyose épidermolytique Naevuspidermolytique Autres Formeskératodermaérythrokératodermie vararabilitésyndrome cutané de pelage ichtyosiforme réticulaire congénitale Syndrome de Maklick | Ichtyose liée à l’X Syndromique Ichtyose réticulaire liée à l’X (syndromique) Ichtyose folliculaire, alopécie et photophobie (IFAP) syndromeconradi – Syndrome de Scammann-happle (chondrodysplasie ponctuelle de type 2) syndromes autosomiques de l’ichtyose de la peau syndromes de l’étherton syndromes de l’hypothrichose syndromes de l’ichtyose-Cholangite sclérosante syndromes de l’hypertrichothystrophyneurologiquesjögren-Larsson syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, retard mental, entéropathie, surdité, neuropathie périphérique, ichtyose, kératodermie.
Seules des données limitées sont disponibles sur l’épidémiologie d’ARCIs. Aux États-Unis, une prévalence à la naissance de 1 pour 100 000 habitants pour LI et de 1 pour 200 000 habitants pour CIE a été estimée. D’autres études ont rapporté une prévalence combinée pour LI et CIE de 1 pour 200 000 à 300 000 habitants.10,11 Dans certains pays comme la Norvège, la prévalence estimée est plus élevée (1 pour 91 000) en raison de mutations fondatrices.12 La découverte d’une ou plusieurs mutations récurrentes dans une population peut être due au fait que la mutation s’est produite à un moment donné de l’histoire et a ensuite été transmise de génération en génération (mutation fondatrice) ou au fait que la région du génome où la mutation est trouvée possède une séquence d’ADN susceptible de mutation (point chaud de mutation). En Espagne, la prévalence estimée de l’ARCI est de 1 pour 138 000 dans la population générale et de 1 pour 61 700 chez les enfants de moins de 10 ans.13 Dans certaines régions d’Espagne, la prévalence pourrait être encore plus élevée. Sur la côte galicienne, par exemple, une prévalence de 1 pour 33 000 a été rapportée, en raison également d’un effet fondateur.14
Ichtyose lamellaire et caractéristiques érythrodermacliniques ichtyosiformes congénitales
Bien que l’on pensait à l’origine que LI et CIE étaient des entités différentes, des cas de patients présentant des manifestations cliniques intermédiaires ont été rapportés et les deux conditions peuvent être causées par des mutations du même gène.15,16 De plus, les patients présentant la même mutation, même au sein d’une même famille, peuvent développer des phénotypes différents.12,15
La plupart des patients naissent enveloppés d’une membrane de collodion qui disparaît progressivement au cours des premières semaines de vie et est remplacée par le phénotype définitif (Fig. 1 BIS). Une hypohidrose, une intolérance à la chaleur sévère et une dystrophie des ongles sont fréquemment observées chez LI et CIE.17-19 Patients atteints de LI présentent généralement des manifestations cliniques plus sévères que ceux atteints de CIE. Ils ont de grandes écailles ressemblant à des plaques, souvent de couleur sombre, couvrant toute la surface du corps. L’érythrodermie est absente ou minime. Ces patients présentent généralement de l’ectropion et parfois de l’éclabium, une hypoplasie du cartilage articulaire et nasal, une alopécie cicatricielle, en particulier au bord du cuir chevelu, et une kératodermie palmoplantaire (Fig. 1B et C). La CIE se caractérise par la présence d’érythrodermie et de fines écailles blanchâtres (Fig. 2). Certains patients présentent un érythème marqué et une mise à l’échelle généralisée. Les écailles peuvent être grandes et de couleur foncée, en particulier sur les surfaces extenseurs des pattes. Dans les cas moins graves, l’érythème est léger et la desquamation est fine.

Caractéristiques cliniques de l’ichtyose lamellaire. A, desquamation lamellaire brunâtre. B, hyperkératose plantaire marquée. C, alopécie cicatricielle du cuir chevelu.
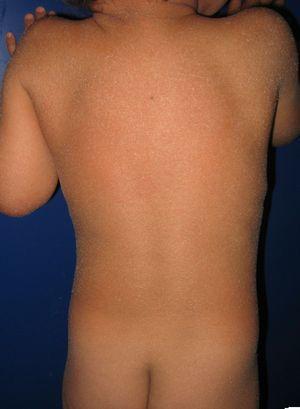
Patient présentant une érythrodermie ichtyosiforme congénitale et des mutations du gène ALOXE3. Un érythème léger et une desquamation furfuracée blanchâtre généralisée peuvent être observés.
Histopathologie
Les modifications histopathologiques ne fournissent pas de diagnostic. Dans le LI, une hyperkératose orthokératotique massive est observée, généralement avec deux fois l’extension comme dans le CIE. L’épiderme est acanthotique et prend parfois une apparence de psoriasis. Le taux de prolifération cellulaire est normal ou légèrement élevé.17-19 Patients atteints de CIE présentent une hyperkératose moins marquée, avec parakératose focale ou étendue, une couche granulaire normale ou épaissie et une acanthose plus prononcée. Le renouvellement épidermique est augmenté.17-19
Ultrastructure
Bien qu’une corrélation étroite entre les résultats moléculaires, cliniques et ultrastructuraux n’ait pas encore été trouvée, la microscopie électronique peut néanmoins être utile pour exclure d’autres formes d’ichtyose et pour guider les analyses génétiques dans certains cas. Quatre types d’ichtyose congénitale ont été décrits (tableau 2).
Classification ultrastructurale des Ichtyoses congénitales.
| Type | Caractéristique principale | Autres Caractéristiques | Mutations | Manifestations cliniques | |
| 1 | Absence de marqueurs ultrastructuraux des ichtyoses de types 2, 3 et 4 | Gouttelettes ou anneaux lipidiques dans la couche cornée (les plus fréquents) De petits granulés de kératohyaline Granules de revêtement membranaire vésiculaire ou lobulaire | TGM1 (33.3%) ALOX12B (2 caisses) | CIE | |
| 2 | Fentes de cholestérol dans la couche cornée | Absence ou amincissement des enveloppes cornéespetits granulats de kératohyaline | Gouttelettes lipidiques | TGM1 (89-100%) | LI |
| 3 | Structures membraneuses stratifiées dans la strate granulosum et / ou la strate corneum. | Granulats de revêtement membranaire anormaux goutteleTtes lipidiques de vacuoles juxtanucléaires proéminentes dans la couche granulaire | NIPAL4 (93%) | CIE (le plus fréquent) LI | |
| 4 | Paquets membranaires trilamellaires qui remplissent certaines cellules de la couche granulosum et/ou de la couche cornée | Granules de revêtement membranaire anormaux | FTAP4 | Syndrome de prématurité de l’ichtyose (100%) |
Abréviations: CIE, érythrodermie ichtyosiforme congénitale; LI, ichtyose lamellaire.
Ichtyose congénitale de type 1
L’ichtyose congénitale de type 1 est caractérisée par l’absence de marqueurs ultrastructuraux pour les ichtyoses de types 2, 3 et 4. Par conséquent, le diagnostic n’est généralement posé que lorsque les autres types ont été exclus. La découverte la plus fréquente est la présence de gouttelettes ou d’anneaux lipidiques dans la couche cornée (Fig. 3 BIS).20 Ces gouttelettes lipidiques ne sont pas une caractéristique constante ou spécifique à ce type particulier car elles ne sont pas présentes dans tous les cas, 20 et elles peuvent être présentes dans d’autres types d’ichtyose.21,22 Cliniquement, la plupart des patients présentent des manifestations de CIE.12,20 Un tiers des patients présentent des mutations du gène TGM1.16 Ce type ultrastructural a également été identifié en association avec des mutations du gène ALOX12B.23,24

Images au microscope électronique. A, ichtyose congénitale de type 1, montrant des gouttelettes lipidiques dans la couche cornée et l’absence de marqueurs ultrastructuraux des autres types d’ichtyose. B, ichtyose congénitale de type 2, caractérisée par la présence de fentes de cholestérol (flèche) dans les cornéocytes.
Ichtyose congénitale de type 2
L’ichtyose congénitale de type 2 est caractérisée par des fentes de cholestérol dans la couche cornée (Fig. 3B).21 De telles fentes sont une constante dans ce type d’ichtyose, et peuvent être détectées dans différentes biopsies chez le même patient; le traitement par rétinoïdes oraux n’a aucun impact sur ces fentes.Des agrégats denses à 12,25 électrons ont également été observés sur des cornéocytes chez certains patients présentant une activité déficiente de la TGase 1.26-28 Cliniquement, la plupart des patients présentent des manifestations sévères de CIE.12 Ce type ultrastructural est fortement associé à des mutations du gène TGM1.12,16
Ichtyose congénitale de type 3
L’ichtyose congénitale de type 3 est caractérisée par des structures membraneuses lamellaires dans la couche granuleuse et / ou la couche cornée. Ces structures sont disposées en bandes autour d’un espace vide proche du noyau.22,29-31 Les manifestations cliniques de ce type sont différentes des autres; l’apparition de l’ichtyose est variable, la desquamation et l’érythème peuvent être parcellaires ou généralisés, et les flexions en particulier sont affectées. Les mutations du gène NIPAL4 sont responsables de 93% des ichtyoses de type 3.32
Ichtyose congénitale de type 4
De manière caractéristique, dans l’ichtyose congénitale de type 4, certaines cellules de la couche granulosum et de la couche cornée sont remplies d’emballages membranaires trilamellaires.33 Ces résultats sont pathognomiques pour le syndrome de prématurité de l’ichtyose, une affection actuellement considérée comme une forme syndromique de l’ichtyose.34,35
Études moléculaires
En termes génétiques, les ARCIs sont très hétérogènes. Le gène TGM1 est associé à la plupart des cas, mais des mutations dans 5 autres gènes (ALOX12B, ALOXE3, NIPAL4, CYP4F22 et ABCA12) ont été rapportées. Fischer et coll.36 a étudié 520 familles avec ARCI et identifié des mutations dans au moins 1 de ces gènes dans 78% des cas (TGM1 dans 32%, NIPAL4 dans 16%, ALOX12B dans 12%, CYP4F22 dans 8%, ALOXE3 dans 5% et ABCA12 dans 5%). Dans une autre étude portant sur 250 patients atteints d’ARCI d’origines différentes, 38% présentaient des mutations TGM1, 6,8% des mutations ALOXE3 et 6,8% des mutations ALOX12B.37 En Galice, nous avons identifié des mutations dans les gènes TGM1, ALOX12B, ALOXE3, NIPAL4 et CYP4F22 dans 75% des familles étudiées, mais la distribution des mutations était différente.14 Le gène TGM1 a été muté en 68.7% des cas alors que le gène ALOXE3 a été muté chez seulement 1 patient. Nous n’avons détecté aucune mutation dans aucun des 3 autres gènes étudiés.
TGM1
Le gène TGM1 est situé sur le chromosome 14q11.2 et possède 15 exons (GenBank NM-000359.2). Il code l’enzyme TGase 1, qui est l’une des 3 enzymes TGase présentes dans l’épiderme.38 Cette enzyme participe à la formation de l’enveloppe cornée en catalysant la réticulation dépendante du calcium de plusieurs protéines telles que l’involucrine, la loricrine et les protéines riches en proline.39,40 Il catalyse également la liaison de ??- hydroxycéramides dans la couche externe de l’enveloppe cornée avec des protéines dans la couche interne.41,42 Chez les patients porteurs de mutations TGM1, l’enveloppe cornée est manquante et l’activité de la TGase 1 est réduite ou inexistante.43-47
Depuis 1995, lorsque ce gène a été identifié comme responsable de certains cas d’ARCI, 48-50 plus de 110 mutations ont été rapportées chez des patients d’origines différentes. Les mutations de TGM1 sont la cause la plus fréquente d’ARCI.36,37 Cette mutation a été retrouvée dans 55% des cas aux États-Unis et dans 84% des cas en Norvège.12,51 La mutation la plus fréquente est c.877-2A > G, qui a été trouvé dans 34% des allèles mutés rapportés à ce jour.52 La fréquence élevée de cette mutation dans des pays tels que les États-Unis et la Norvège est due à un effet fondateur.12,53 La deuxième mutation la plus fréquente est p. Arg142His. Cette mutation et des mutations similaires ont été rapportées dans des pays tels que l’Égypte, l’Allemagne, la Finlande et les États-Unis, 15, 49-51, 54-56 et il semblerait qu’il s’agisse de mutations à points chauds.57 La mutation p. Arg307Trp est fréquente dans la population japonaise.5 En Galice, le p. Arg760X, c. 1223_1227delACACA et c.984 +1G > Mutations A dans TGM1 ont été identifiées dans 81,82% des familles avec des mutations dans ce gène, suggérant un effet fondateur.14 La confirmation de cette hypothèse a été obtenue par étude de l’haplotype (travaux encore inédits).
Les mutations TGM1 sont responsables de la plupart des cas de LI15,27,44,46,56,58-63 et pour un petit pourcentage de cas de CIE.43,47,64,65 De telles mutations peuvent également donner lieu à d’autres formes d’ARCI telles que SHCB, SHCB acral et ichtyose en maillot de bain.
De nombreuses études ont tenté de démontrer des associations génotype-phénotype entre des mutations de TGM1 et des résultats ultrastructuraux ou cliniques, mais aucune corrélation significative n’a été observée à ce jour.15,16,53 En général, les patients présentant des mutations du gène TGM1 sont plus gravement touchés que ceux qui n’en ont pas. Dans une étude portant sur 83 patients atteints d’ARCI en Suède et en Estonie, la présence d’ectropion et de collodion baby a été associée à des mutations de TGM1, tandis qu’un taux plus élevé d’érythème a été observé chez les patients sans mutation de ce gène.66 Une autre étude a montré que le type de mise à l’échelle est la principale différence entre les porteurs et les non porteurs de mutations TGM1, en constatant que tous les patients présentant des mutations dans ce gène présentaient une mise à l’échelle lamellaire alors que 80% de ceux sans mutation TGM1 présentaient une mise à l’échelle fine.14 En outre, il a été constaté que les mutations tronquées sont plus fréquemment associées à une hypohidrose et à des troubles de la transpiration que les mutations faux sens.51 Dans la population nord-américaine, un modèle basé sur la présence de certaines caractéristiques cliniques prédit que les patients nés en tant que bébés collodions et présentant des troubles oculaires et / ou une alopécie sont 4 fois plus susceptibles d’avoir des mutations TGM1.51
ALOXE3 et ALOX12B
Les gènes ALOXE3 et ALOX12B sont situés sur le chromosome 17p13.1.67 Ils ont une structure similaire avec 15 exons qui codent les LOX épidermiques eLOX-3 et 12R-LOX.68,69 Le fait qu’ils soient majoritairement exprimés dans les couches suprabasales de l’épiderme soutient leur rôle dans les phases avancées de différenciation épidermique, avec participation au traitement des corps lamellaires.24,70 Ces enzymes agissent sur les étapes adjacentes de la voie de l’hépoxiline (Fig. 4). Le 12R-LOX transforme l’acide arachidonique en acide 12R-hydroxyeicosatétraénoïque tandis que l’eLOX-3 convertit ce produit en un isomère époxyalcool 69,71 de la famille de l’hépoxiline A3.72 Le produit de l’hépoxiline est instable et est hydrolysé dans les cellules en un dérivé trihydroxy spécifique (trioxiline). Bien que le rôle exact des produits de la voie de l’hépoxiline ne soit pas connu, il a été émis l’hypothèse qu’ils pourraient participer à la formation de lipides intercellulaires de la couche cornée ou agir comme des signaux induisant la différenciation des kératinocytes.
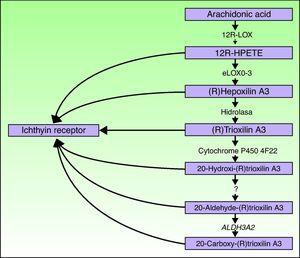
Schéma de la voie de l’hépoxiline, montrant la participation des gènes ALOXE3, ALOX12B, NIPAL4 et CYP4F22. Des mutations dans ces gènes sont responsables de certains types d’ARCI. HPETE indique l’acide hydroperoxyeicosatétraénoïque.
Les gènes ALOX12B et ALOXE3 ont été identifiés pour la première fois en 200273,74 Depuis lors, plus de 30 mutations dans le gène ALOX12B23,24,37, 75-77 et environ 10 dans le gène ALOXE337,74,75 ont été rapportées. Ces mutations sont responsables de 14% à 17% des ARCIs36, 37 et 72.2% des CSH.23,78,79 La relation causale entre ces mutations et le phénotype a été confirmée en démontrant que l’activité catalytique de la LOX épidermique était totalement abolie chez les patients atteints de ces mutations75,80 et en utilisant des modèles animaux reproduisant le phénotype ichtyosiforme observé chez l’homme.81-83 Les deux gènes sont responsables d’un pourcentage similaire de cas d’ARCI. Cependant, la gamme des différentes mutations du gène ALOXE3 est limitée, en raison de la prédominance de 2 mutations, p. Arg234X et p. Pro630Leu, qui semblent correspondre à des points chauds.37,74,75
Les patients présentant des mutations dans les gènes ALOXE3 et ALOX12B présentent généralement un phénotype CIE.74,75,77 La gravité de l’écaillage est légère ou modérée et les écailles ont une couleur blanchâtre ou brun clair. Un érythème peut également être présent. Jusqu’à 76% des patients sont nés en tant que bébés collodions et 88% ont des troubles de la transpiration.37 Patients présentant des mutations du gène ALOX12B présentent une desquamation blanchâtre plus limitée que les porteurs de mutations du gène ALOXE3. Dans ces cas, les écailles sont brunâtres et adhérentes. La présence d’érythème, d’hyperkératose palmoplantaire et l’accentuation des plis palmoplantaires sont également associées à des mutations ALOX12B.37
Ichtyine/NIPAL4
Le gène NIPAL4, également connu sous le nom de gène ichtyine, est situé sur le chromosome 5q33. Il possède 6 exons qui codent une protéine avec plusieurs domaines transmembranaires de fonction inconnue.84 On a émis l’hypothèse que le produit protéique participe à la même voie métabolique que LOX et pourrait agir comme récepteur des trioxilines A3 et B3 ou d’autres métabolites de la voie métabolique de l’hépoxiline.84 Il serait ainsi impliqué dans la formation des corps lamellaires ou dans leur transport vers l’espace extracellulaire.32 À l’appui de cela, 2 observations. Premièrement, dans 93% des cas, les mutations de ce gène sont associées à un schéma ultrastructural d’ichtyose congénitale de type 3, caractérisé par des anomalies dans les corps lamellaires et la présence de membranes périnucléaires allongées dans la strate granulosum.32 Deuxièmement, NIPAL4 s’exprime essentiellement dans la strate granulosum de l’épiderme, où sont présents les corps lamellaires.85
Depuis la découverte du gène NIPAL4 en 200484, seules 9 mutations ont été rapportées chez des patients des pays méditerranéens (Algérie, Turquie et Syrie), 84 pays scandinaves, 32 Pakistan, 85 îles Féroé, 32 et Amérique du Sud.84
Le spectre clinique des patients présentant des mutations de ce gène est large, même parmi les membres d’une même famille. Entre 3,7% 32 et 60% 84 naissent en tant que bébés collodions. Lorsque la membrane du collodion disparaît, la plupart des patients développent des manifestations de CIE, avec de fines écailles blanchâtres sur une base érythémateuse sur le visage et le tronc et de plus grandes écailles brunâtres sur le cou, les fesses et les jambes.84 Une xérose marquée, des plaques hyperkératotiques réticulaires brunâtres généralisées apparaissant accentuées dans les plis cutanés et une dyschromie faciale peuvent être présentes.32,85 De plus, la kératodermie palmoplantaire est une découverte fréquente avec des contractures occasionnelles des doigts et des ongles incurvés. Certaines études ont rapporté des résultats plus typiques de LI.32,85 La présence de signes et de symptômes de dermatite atopique a été rapportée chez certains patients, bien que des mutations du gène FLG n’aient été détectées dans aucun de ces cas.85
CYP4F22
Le gène FLJ39501 ou CYP4F22 est situé sur le chromosome 19p13.12.86 Il possède 12 exons87 et code pour un cytochrome P450, famille 4, sous-famille F, polypeptide 2, homologue de la leucotriène B4-ω-hydroxylase (CYP4F2). La réaction catalysée par le produit de FLJ39501 dans la peau et les substrats de cette réaction peut être déduite par analogie avec ses homologues connus CYP4F2 et CYP4F3.88 On a émis l’hypothèse que le CYP4F2 et le CYP4F3 participent à la voie de l’hépoxiline en catalysant la conversion de la trioxiline A3 en 20-hydroxy-(R)trioxiline A387 et que le produit final de cette voie, la 20-carboxy-trioxiline A3, pourrait avoir un effet régulateur biologique clé sur la peau.89
À ce jour, seules 8 mutations de ce gène ont été rapportées dans 12 familles consanguines de pays méditerranéens87 et dans 1 famille d’origine israélienne.62
Dans les familles rapportées par Lefèvre et al., 87 la plupart des patients avaient un phénotype CIE à la naissance et celui-ci a ensuite progressé vers LI. Les patients sont généralement nés avec une érythrodermie marquée, mais sans membrane de collodion. En vieillissant, ils ont développé une desquamation gris blanchâtre généralisée, plus marquée dans la région périumbilicale, sur les fesses et sur la partie inférieure du corps. L’hyperlinéarité des paumes et des plantes et la desquamation du cuir chevelu, parfois de type pityriasiforme, étaient fréquentes.87 Dans une autre famille, les 3 membres touchés sont nés en tant que bébés collidions et ont développé une érythrodermie intense, une desquamation généralisée et une kératodermie palmoplantaire.62
ABCA12
En 2003, le gène ABCA12 a été signalé comme responsable de certains cas de LI et a été cartographié sur le chromosome 2q34.4 Il a ensuite été confirmé que des mutations de ce gène étaient également responsables de HI.2, 3ABCA12 code pour 53 exons et appartient à une famille de transporteurs ABC, qui lient l’adénosine triphosphate tout en facilitant le transport de plusieurs molécules à travers la membrane cellulaire.90 Les membres de la sous-famille ABCA sont tous impliqués dans le transport des lipides.91 Une fonction ABCA12 déficiente provoque des troubles du transport des lipides dans les corps lamellaires et entraîne donc une diminution des taux de lipides intercellulaires dans la couche cornée.3 Des études ultrastructurales ont montré que l’ABCA12 est situé dans les corps lamellaires associés aux glycosylcéramides.Les mutations de 91ABCA12 ont été associées à des troubles de la distribution et du transport des glycosylcéramides et à une diminution des taux d’hydroxycéramides, l’un des principaux composants de la barrière lipidique dans les espaces intercellulaires.3,6,92,93 L’hyperkératose massive qui survient chez ces patients pourrait être une réponse compensatoire à une barrière lipidique déficiente.94 Elle pourrait également être due à l’absence de desquamation des cornéocytes, 93 qui pourrait être causée par des défauts de transport de certaines protéases, telles que la callicréine 5 et la cathepsine D, résultant de troubles des corps lamellaires.95 modèles murins et études in vitro suggèrent que les mutations ABCA12 ont également un effet sur la différenciation épidermique.95-97
À ce jour, plus de 50 mutations du gène ABCA12 ont été rapportées chez des patients atteints d’ARCI d’Afrique, d’Europe, du Pakistan et du Japon. Les mutations les plus fréquentes sont p. Val244SerfsTer28, 2, 98, 99 identifiées dans les populations pakistanaises et indiennes, et p. Asn1380Ser,4 identifiées dans les familles africaines. Dans les deux cas, il peut s’agir de mutations fondatrices.
L’étendue des mutations ABCA12 est liée au phénotype, les mutations associées à une perte complète de la fonction conduisant au phénotype HI.2,3,98-102 En revanche, chez LI et CIE, la plupart des mutations sont fausses et ont un effet moins sévère sur la fonction protéique.4-6,103 Les mutations sous-jacentes au phénotype LI semblent être concentrées dans la première région de cassette de liaison de l’adénosine triphosphate.4 Cliniquement, les patients atteints de CIE et de mutations du gène ABCA12 présentent des écailles de taille moyenne un peu plus grandes que celles habituellement observées chez les patients atteints de ce phénotype.
Ichtyose arlequine
HI ou fœtus arlequin est une forme sévère et généralement fatale d’ichtyose. Les enfants sont généralement prématurés avec de vastes plaques hyperkératotiques brillantes, séparées par des fissures profondes, qui recouvrent tout le tégument et forment des motifs géométriques rappelant les vêtements portés par les arlequins, donnant ainsi son nom à la condition. L’oppression cutanée entraîne une éversion marquée des paupières et des lèvres, un développement rudimentaire du cartilage articulaire et nasal et, occasionnellement, une microcéphalie. Les enfants ont rarement des cils ou des sourcils, bien que les cheveux du cuir chevelu puissent être conservés. Les mains et les pieds sont enflés et œdémateux, et souvent recouverts d’une couche semblable à un gant. Ils peuvent avoir des contractures des doigts.
Pour ces patients, le risque de mourir pendant la période néonatale est très élevé.104 La ventilation pulmonaire est compromise; la perte d’eau transépidermique entraîne une déshydratation, un déséquilibre hydroélectrique et une instabilité thermique; et le risque d’infections est accru. L’oppression faciale et l’éclabium entravent la succion et donc l’alimentation, avec l’aggravation correspondante de la déshydratation. Les nouveau-nés atteints de cette maladie vivaient rarement plus longtemps quelques semaines. Ces dernières années, cependant, les chances de survie à long terme ont considérablement augmenté, essentiellement en raison de l’administration de rétinoïdes systémiques et des progrès des soins néonatals intensifs.105 Dans une étude récente, 83% des patients traités par rétinoïdes oraux ont survécu contre 24% des patients non traités. La plupart des décès sont survenus au cours des 3 premiers jours de la vie, mais le traitement n’a commencé qu’après cela chez de nombreux survivants.104 Cela suggère que bon nombre de ces décès précoces se seraient produits indépendamment du traitement aux rétinoïdes.
Les enfants qui survivent à la période néonatale développent généralement une CIE sévère.106 La nature et la localisation des mutations du gène ABCA12 et l’étendue de la perte de la fonction de transport peuvent déterminer le pronostic.3,92,107 Patients qui conservent un certain degré d’activité protéique, bien que minime, peuvent avoir de meilleures chances de survivre. Les porteurs de mutations homozygotes ont un taux de mortalité plus élevé.104
La principale caractéristique histologique de HI est la présence d’une couche cornée orthokératotique extrêmement épaisse et compacte. Les follicules pileux et les canaux sudoripares ont des bouchons hyperkératosiques107,108 proéminents et présentent des corps lamellaires anormaux ou absents, des inclusions lipidiques ou des restes d’organites ou de noyaux dans les cornéocytes, et l’absence de lipides intercellulaires dans l’étude ultrastructurale.108,109 Les follicules pileux présentent une concentration marquée de matériel kératotique, qui est une caractéristique diagnostique de HI utilisée pour le diagnostic prénatal.
À ce jour, le taux de détection des mutations du gène ABCA12 chez les patients atteints de HI est proche de 100%, ce qui semble être une condition génétiquement homogène.
Bébé Collodion et Bébé Collodion Auto-cicatrisant
Les bébés collodions naissent généralement prématurément et la morbidité et la mortalité périnatales augmentent. À la naissance, le nouveau-né est recouvert d’une membrane transparente brillante rappelant l’emballage en cellophane (Fig. 5). Les bébés ont de l’ectropion, de l’éclabium et une hypoplasie du cartilage nasal et articulaire. La succion et la ventilation pulmonaire peuvent être entravées110 et la perte transépidermique d’eau et le risque d’infections sont augmentés.110,111

Bébé collodion qui a ensuite évolué vers un phénotype d’ichtyose lamellaire.
Collodion baby est la présentation habituelle de HI and CIE. LI autosomique dominante, 112, 113 Syndrome de Sjögren-Larsson, 110 trichothyodystrophie, 114 maladie de Gaucher juvénile, 110 maladie du stockage des lipides neutres, syndrome de Conradi-Hünermann-Happle, syndrome de Hays-Wells et dysplasie ectodermique115 peuvent également se présenter occasionnellement en tant que bébé collodion. La membrane disparaît spontanément chez 10% à 24% des nouveau-nés, pour laisser place à une peau tout à fait normale.110 116 Dans le passé, ces cas étaient décrits comme des cas de LI du nouveau-né,117 mais ils ne sont pas désignés sous le nom de SHCB.118 Certains auteurs ont suggéré le terme d’ichtyose au collodion auto-améliorant parce que beaucoup de ces patients, lorsqu’ils sont réexaminés plus tard dans l’enfance ou à l’âge adulte, présentent un degré variable d’anhidrose et d’intolérance à la chaleur et de légers signes d’ichtyose, tels que xérose et desquamation fine, en particulier dans les aisselles et le cou.78
Ni la microscopie optique ni les études ultrastructurales du bébé collodion ne sont spécifiques. Il est donc préférable de retarder la biopsie cutanée jusqu’au développement du phénotype définitif.
Des mutations dans les gènes TGM1, 7, 119ALOXE3,78 et ALOX12B23,78,79 ont été identifiées chez des patients atteints de SHCB. Les mutations ALOX12B sont les plus courantes. Dans une série de 15 patients scandinaves atteints de SHCB, 67% présentaient des mutations dans le gène ALOX12B, 25% dans le gène ALOXE3 et 8,3% dans le gène TGM1.78 Mutations n’ont pas été trouvées chez certains patients, et d’autres gènes sont donc également susceptibles d’être impliqués. Il a été spéculé que ces mutations réduisent l’activité enzymatique dans l’utérus, mais pas après la naissance.7 Dans l’utérus, où la pression hydrostatique est élevée, la chélation par l’eau convertit l’enzyme mutée en une conformation inactive. Après la naissance, lorsque la pression diminue, l’enzyme reprend sa forme active et son activité augmente suffisamment pour maintenir un phénotype normal ou peu affecté.7
Bébé Collodion auto-cicatrisant Acral
Bien que le bébé collodion affecte tout le corps, des cas confinés aux régions acrales ont été rapportés. En 1952, Finlay et al.120 a signalé un cas de membrane de collodion qui n’affectait que les mains et les pieds et qui suivait un cours d’auto-guérison. Récemment, un nouveau cas de SHCB acral a été rapporté en association avec des mutations du gène TGM1.8 On ne sait pas pourquoi ces lésions sont limitées aux régions acrales, bien que des facteurs associés à la régulation de l’activité enzymatique dépendante du site puissent fonctionner.8
Ichtyose du maillot de bain
L’ichtyose du maillot de bain a été signalée pour la première fois en tant que variante ARCI indépendante en 2005, bien que des cas d’ichtyose avec une distribution particulière aient été signalés précédemment.121-123 Il a été détecté principalement chez des patients d’origine sud-afrique9, bien qu’il ait également été signalé chez des individus d’Europe et de pays méditerranéens.124 À la naissance, les patients ont une membrane de collodion généralisée qui se détache ensuite pour laisser la distribution caractéristique de l’écaillage. Le tronc, la région proximale des bras, y compris les aisselles, le cou et le cuir chevelu sont généralement touchés, tandis que la partie centrale du visage, les membres et la région surrénale sont généralement épargnés.9 Les écailles sont grandes, lamellaires et de couleur sombre. Une desquamation plus fine peut survenir dans les fosses poplitées et antécubitales.124,125 Les paumes des mains et la plante des pieds présentent une légère hyperkératose diffuse alors que le dos des mains et des pieds ne présente aucune atteinte.
L’étude histopathologique de la peau affectée montre une hyperkératose marquée sans parakératose, des couches granulaires normales, une acanthose légère ou modérée et un infiltrat lymphocytaire léger dans le derme supérieur.9 Les observations en microscopie électronique concordent dans la plupart des cas avec l’ichtyose congénitale de type 2. La peau non impliquée ne présente aucun résultat anormal.124,125 Dans une peau saine, l’activité de la TGase 1 est légèrement réduite et généralement localisée dans les zones péricellulaires. Dans la peau impliquée, l’activité enzymatique est résiduelle et anormalement située dans le cytoplasme.124 Mutations
ont été détectées dans le gène TGM1 chez tous les patients atteints d’ichtyose du maillot de bain étudiés à ce jour.119,124-126 La mutation la plus fréquente est p. Arg315Leu, qui a été identifiée chez la plupart des patients sud-africains et pourrait être une mutation fondatrice. Oji et coll.124 a suggéré que la température de la peau pourrait jouer un rôle dans le développement de ces manifestations. En utilisant la thermographie numérique, les auteurs ont montré une forte corrélation entre la température corporelle et la desquamation, les zones les plus chaudes du corps étant les plus touchées. Aufenvenne et coll.127 a montré une diminution de la température optimale pour l’activité de la TGase 1 chez les patients atteints d’ichtyose en maillot de bain. Cette diminution n’a pas été observée chez les témoins sains ni chez les patients présentant une LI généralisée. Cette diminution de la température expliquerait le phénotype de ces patients. La température optimale est de 37°C pour l’enzyme normale mais de 31°C pour l’enzyme mutée.
Traitement
L’objectif principal du traitement de l’ichtyose est d’éliminer le tartre et de réduire la xérose sans provoquer d’irritation excessive (Tableau 3). Avant de décider du traitement, des aspects tels que l’âge et le sexe du patient, le type et la gravité de la maladie, ainsi que l’étendue et le site des lésions doivent être pris en compte.128
Stratégie thérapeutique dans les Ichtyoses congénitales Autosomiques récessives.
| Stratégie thérapeutique pour les ichtyoses congénitales autosomiques récessives | |
| Bain et élimination mécanique des écailles | Bain avec du bicarbonate de sodium ou de l’amidon de blé, de maïs ou de riz; enlèvement mécanique des écailles (1 ou 2 fois par jour) |
| Traitement topique (séquentiel) | Hydratants contenant de l’uréecératinolytiques avec du propylèneglycolcombiné kératinolytiques (propylèneglycol, acides α-hydroxy ou urée) Kératinolytiques combinés avec de l’acide salicyliquetopical Rétinoidsdans les nouveau-nés et les jeunes enfants, appliquez un véhicule sans ingrédients actifs. Évitez l’urée, l’acide salicylique et l’acide lactique en raison du risque d’absorption systémique |
| Traitement oral | Rétinoïdes oraux (acitrétine ou isotrétinoïne) |
| Autres mesures | Suivi de l’ectropion par l’ophtalmologiste Nettoyage régulier de l’oreille externe par le spécialiste oreille-gorge-nez Physiothérapie pour prévenir les contractures.Eviter les activités pénibles à haute température ambiantehydrothérapie |
Bain et Élimination mécanique des écailles
Un bain quotidien est recommandé pour les patients atteints d’ARCI afin d’éliminer mécaniquement les écailles et les traces de crème hydratante. C’est plus facile si le patient est immergé dans l’eau pendant 15 à 30 minutes. Certains auteurs recommandent d’ajouter du bicarbonate de sodium au bain pour dénaturaliser les kératines et rendre l’eau alcaline, et ainsi faciliter l’élimination des écailles.129 Les autres produits pouvant être ajoutés comprennent l’amidon de blé, l’amidon de maïs ou l’amidon de riz. Les huiles de bain ne sont pas appropriées car elles peuvent entraîner une occlusion avec un risque ultérieur de prolifération bactérienne et d’aggravation de la thermorégulation.
Traitement topique
Les hydratants et les agents kératolytiques topiques sont généralement la première option thérapeutique. Ils améliorent la fonction barrière cutanée et facilitent la desquamation. Des effets indésirables locaux légers, tels qu’un prurit transitoire, une irritation ou une sensation de picotement peuvent survenir.
Le chlorure de sodium, l’urée, l’acétate de vitamine E, le glycérol et la vaseline peuvent être utilisés comme hydratants et lubrifiants. Chez les patients présentant une desquamation épaisse et une hyperkératose marquée, 1 ou plusieurs agents kératolytiques, tels que les acides α-hydroxy (acide lactique et glycolique), 130 acide salicylique, la N-acétylcystéine, 131-133 urée (> 5%), 134 et le propylène glycol, peuvent être ajoutés. Des modulateurs de différenciation des kératinocytes sont également utilisés. Ceux-ci comprennent les rétinoïdes topiques (trétinoïne, adapalène, tazarotène), le calcipotriol 135, 136, le 137 et le dexpanthénol.Les rétinoïdes topiques provoquent souvent des irritations et de petites fissures très douloureuses.137 De plus, il existe un risque d’absorption et de tératogénicité chez les femmes fertiles si elles sont trop utilisées.138 Pour améliorer l’efficacité des kératolytiques et des hydratants, un pansement occlusif peut être appliqué dans des zones spécifiques réfractaires au traitement.139 Un effet additif ou synergique peut également être obtenu en combinant 2 agents kératolytiques ou hydratants ou plus.140-142 Le traitement doit être optimisé pour chaque individu, compte tenu de la nature très variable de la maladie et de la sensibilité de la peau et des différences de réponse à chaque traitement. Le processus d’optimisation peut être aidé en traitant un côté du corps différemment de l’autre pour permettre des comparaisons. Les nouveau-nés et les jeunes enfants doivent être traités avec un véhicule sans aucune substance active car la peau est très fine et sensible et la plupart des kératolytiques ne sont pas tolérés. De plus, le risque d’absorption percutanée de produits topiques tels que l’urée, l’acide salicylique et l’acide lactique est plus élevé.143-145
Traitement systémique
Les rétinoïdes oraux ont des effets kératolytiques qui aident à éliminer les écailles et à prévenir l’hyperkératose excessive. L’isotrétinoïne et les rétinoïdes aromatiques (acitrétine et étrétinate) se sont révélés efficaces dans le traitement de l’ARCIs.128 146 147 L’acitrétine à une dose de 0,5 à 1 mg / kg / j est le médicament le plus utilisé, en particulier chez les patients atteints de LI. 148 Les patients atteints de CIE peuvent avoir une réponse plus complète et à des doses plus faibles.
Les principaux effets indésirables sont les troubles cutanés, la tératogénicité, les troubles musculo-squelettiques et le profil lipidique anormal et l’élévation des transaminases.149-152 En ce qui concerne la tératogénicité, dans le cas de l’étrétinate et de l’acitrétine, les médicaments doivent être évités pendant la grossesse et les patientes doivent éviter de tomber enceintes pendant 3 ans après l’arrêt du traitement.L’isotrétinoïne 151 a une demi-vie plus courte et est complètement éliminée de l’organisme après 1 mois et peut donc être l’option préférée chez les femmes qui souhaitent devenir enceintes.128
La surveillance du traitement doit inclure un bilan de laboratoire avec un test de la fonction hépatique et le profil lipidique avant de commencer le traitement, puis à 1 mois et tous les 3 mois après le début du traitement. Chez les femmes fertiles, un test de grossesse doit être effectué dans les 2 semaines précédant le début du traitement et une mesure contraceptive efficace doit être utilisée 4 semaines avant le traitement jusqu’à 3 ans après (dans le cas de l’acitrétine). Lorsqu’un traitement prolongé par des rétinoïdes est nécessaire, la croissance et le développement osseux doivent être surveillés. Certains auteurs suggèrent de réaliser une étude osseuse avant le traitement suivie d’un examen annuel.151 Les directives récentes ne recommandent pas d’effectuer une radiographie de routine en raison des effets nocifs possibles.152 Au lieu de cela, des études radiographiques sélectives sont recommandées chez les patients souffrant de douleurs osseuses atypiques.152
Une alternative au traitement systémique des rétinoïdes est l’utilisation de médicaments appelés agents bloquant le métabolisme de l’acide rétinoïque, qui augmentent les niveaux endogènes d’acide rétinoïque. L’un de ces médicaments est le liarozole, qui a obtenu le statut d’orphelin pour le traitement de LI, CIE et HI par l’Agence européenne des médicaments et la Food and Drug Administration américaine.153-155 Ce médicament s’est avéré plus efficace que l’acitrétine dans les essais cliniques et il est également mieux toléré et a un meilleur profil pharmacocinétique.154
Autres Soins médicaux
Chez les patients atteints d’ectropion, l’application de larmes artificielles et de lubrifiants oculaires et l’hydratation de la peau du visage et des joues en particulier peuvent réduire la rétraction palpébrale. La correction chirurgicale est une option valable dans les cas graves, mais elle doit généralement être répétée quelques années plus tard. L’hydrothérapie peut être bénéfique.156 Patients doivent être invités à éviter une activité physique intense lorsque la température ambiante est élevée, étant donné que l’hypohidrose entraîne un risque de coup de chaleur et de convulsions. Les rétinoïdes oraux peuvent améliorer la thermorégulation.157 La physiothérapie est importante pour prévenir la contracture de flexion, en particulier dans le cas de HI. Un nettoyage régulier du conduit auditif externe par un spécialiste de l’oreille, de la gorge et du nez peut empêcher l’accumulation d’écailles et ainsi prévenir la perte auditive.
Conseil génétique et diagnostic prénatal
Lorsqu’un patient reçoit un diagnostic d’ichtyose, il doit se voir proposer un conseil génétique approprié dans lequel la nature du trouble, le mode de transmission et le risque de manifestations futures dans la famille sont expliqués. Le diagnostic prénatal peut indiquer si le fœtus est affecté et, si tel est le cas, une préparation psychologique de la famille peut être proposée et des problèmes anticipés pendant la grossesse et l’accouchement. Les parents peuvent avoir la possibilité d’un avortement si aucun traitement n’est disponible. De plus, si la thérapie génique pour ces affections devenait disponible à l’avenir, le diagnostic prénatal permettrait l’application de cette thérapie le plus tôt possible.
Pendant plus de 20 ans, le diagnostic prénatal a été réalisé en prélevant un échantillon de biopsie de la peau fœtale et en l’étudiant par microscopie optique, microscopie électronique ou immunohistochimie.158 159 Cette procédure invasive ne pouvait être pratiquée que dans les phases tardives de la grossesse, entre les semaines 15 et 23 de la gestation, et était associée à un risque de 1% à 3% de perdre le fœtus.160,161 L’identification des mécanismes moléculaires des troubles cutanés héréditaires a permis un diagnostic beaucoup plus précoce basé sur des techniques génétiques.L’ADN fœtal 102 162-164 est obtenu par amniocentèse réalisée entre les semaines 15 et 20 ou par prélèvement de villosités choriales entre les semaines 10 et 12. Le risque de perte fœtale avec ces techniques est inférieur à entre 0,5% et 1%.165 D’autres méthodes non invasives en cours de développement sont l’analyse de l’ADN cellulaire fœtal et de l’ADN fœtal libre en circulation maternelle166 ainsi que l’utilisation d’ultrasons tridimensionnels.167,168
Le diagnostic génétique préimplantatoire pourrait également être possible dans les techniques de fécondation in vitro, de sorte que seuls les ovules fécondés exempts de la mutation sont implantés dans l’utérus, évitant ainsi la nécessité d’un avortement dans la plupart des cas.169
Stratégies futures pour le Traitement génétique de l’ichtyose
Bien que des progrès importants aient été réalisés dans le diagnostic génétique de l’ichtyose, de nouvelles stratégies sont également mises en œuvre pour ces maladies.170 La peau étant l’organe le plus accessible pour les thérapies de transfert de gènes, ces techniques sont peu invasives.171 Cependant, la peau présente également des caractéristiques immunologiques uniques qui ne favorisent pas l’expression à long terme d’un produit transgénique.172 Dans LI, un processus de transfert de gènes ex vivo a réussi à restaurer l’expression normale de TGM1 et à corriger le phénotype de peau transplantée sur le dos de souris immunodéprimées.173 174 Récemment, le phénotype des kératinocytes en culture de patients atteints de HI dû à des mutations du gène ABCA12 a également été retrouvé.3
Conflits d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts.