La génétique de la fente labiale et palatine
 Intro / Abstractla lèvre gauche avec ou sans fente palatine est une anomalie congénitale complexe qui peut être isolée ou observée avec d’autres malformations. Il peut également faire partie du phénotype d’un syndrome génétique. Cet article sert d’examen de la prévalence de la fente labiale et palatine, des risques de récidive et des risques d’autres anomalies congénitales. Les syndromes génétiques et les expositions tératogènes connus pour être associés aux fentes orales seront explorés. De plus, les tests génétiques couramment demandés dans le cadre de la génétique clinique pédiatrique pour l’évaluation du patient avec une fente labiale et palatine seront discutés.
Intro / Abstractla lèvre gauche avec ou sans fente palatine est une anomalie congénitale complexe qui peut être isolée ou observée avec d’autres malformations. Il peut également faire partie du phénotype d’un syndrome génétique. Cet article sert d’examen de la prévalence de la fente labiale et palatine, des risques de récidive et des risques d’autres anomalies congénitales. Les syndromes génétiques et les expositions tératogènes connus pour être associés aux fentes orales seront explorés. De plus, les tests génétiques couramment demandés dans le cadre de la génétique clinique pédiatrique pour l’évaluation du patient avec une fente labiale et palatine seront discutés.
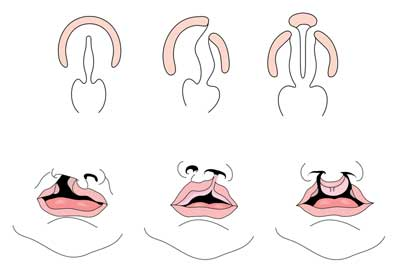
La fente labiale avec ou sans fente palatine (CL/CP) diffère d’une fente palatine isolée (CP) aux niveaux embryonnaire, épidémiologique et génétique. La fente labiale résulte généralement du fait que la proéminence maxillaire et la proéminence nasale médiale ne fusionnent pas entre la cinquième et la sixième semaine de développement embryonnaire. Le développement normal du palais résulte de la formation du palais primaire et du palais secondaire. Le palais primaire est formé aux semaines six à sept par le développement et la fusion des processus nasaux médiaux, nasaux latéraux et maxillaires. Le palais secondaire provient des tablettes palatales (qui se développent à partir des processus maxillaires appariés de la première arcade branchiale) devenant horizontal et fusionnant, formant les palais durs et mous vers la neuvième semaine de développement embryonnaire. Les étagères fusionnent également avec le palais primaire et la cloison nasale. (1)
Les fentes buccales sont l’une des malformations congénitales les plus courantes observées en pépinière néonatale, avec une prévalence globale de 1.6 pour mille nouveau-nés dans le monde, avec CL / CP observé dans environ un pour mille naissances et CP observé dans 0,6 pour mille naissances. (2) Il y a une fréquence plus élevée de CL / CP chez les individus d’origine asiatique, africaine et amérindienne. La CL / CP est également plus fréquente chez les hommes. En revanche, il n’y a pas de différence significative dans l’incidence de la CP entre les différentes origines ethniques, et la CP est plus fréquente chez les femmes. (3) Les risques de récidive au sein d’une famille dépendent du fait que la fente soit isolée (sans autres résultats cliniques) ou considérée comme faisant partie d’un syndrome génétique. La plupart des cas de fentes orales sont isolés (environ 80%). On pense que les fentes isolées ont une hérédité multifactorielle: elles sont dues à une combinaison de plusieurs facteurs, à la fois génétiques et environnementaux. Le risque de récidive (tableau 1) augmente lorsqu’il y a plus d’un parent affecté. Le risque de récidive augmente également plus le défaut est grave.

Une fente labiale et palatine peut être observée avec d’autres anomalies congénitales. La probabilité d’une étiologie génétique ou tératogène augmente le nombre d’anomalies congénitales avec lesquelles un patient se présente. La présence d’autres problèmes tels que la déficience intellectuelle, des problèmes de comportement tels que l’autisme, des caractéristiques dysmorphiques ou d’autres problèmes médicaux rendra également plus probable un trouble génétique ou une exposition tératogène. Environ 13% des personnes atteintes de fente labiale auront d’autres problèmes médicaux ou anomalies. Le nombre augmente à 37% avec la fente labiale et palatine et à 47% avec la fente palatine seule.
Il a été démontré que l’exposition prénatale à des agents tératogènes (tels que la thalidomide, les anticonvulsivants, l’alcool, l’acide rétinoïque et les cigarettes) et aux maladies maternelles (telles que le diabète, la rubéole et la carence en folates) augmentaient le risque de fentes orales. La présence de bandes amniotiques augmente également le risque de fentes. La supplémentation en acide folique périconceptuel est connue pour réduire le risque de fentes orales.
La séquence de Pierre Robin est une anomalie craniofaciale caractérisée par une hypoplasie ou une micrognathie mandibulaire, une fente palatine secondaire en forme de U et une glossoptose entraînant une apnée obstructive et des difficultés d’alimentation. La séquence de Pierre Robin peut être considérée comme faisant partie des syndromes génétiques (syndrome de délétion 22q11.2, syndrome de Stickler; décrit ci-dessous). (5)
Il existe des centaines de syndromes génétiques associés aux fentes buccales, y compris des anomalies cytogénétiques (aneuploïdies, microdélétions) et des troubles monogéniques (Mendéliens). La confirmation d’un diagnostic génétique est essentielle pour déterminer le pronostic et établir un risque de récidive.
Les aneuploïdies telles que la trisomie 13 et 18 ont une forte association avec la CL / CP. La trisomie 13 (syndrome de Patau) est associée à trois copies du chromosome 13, ou translocations robertsoniennes déséquilibrées impliquant le chromosome 13. Les bébés nés avec cette maladie meurent généralement au cours de la période néonatale. Les caractéristiques cliniques comprennent une fente labiale et palatine, un retard de croissance, des malformations graves du système nerveux central (y compris l’holoprosencéphalie), une microcéphalie, une microptalmie, un colobome de l’iris, une absence des yeux, des oreilles malformées, une polydactylie, des poings serrés, des pieds à bascule, des malformations cardiaques congénitales et des malformations urogénitales. Des fentes de la ligne médiane (sinon très rares) peuvent être observées dans la trisomie 13 en raison du risque de défauts de la ligne médiane, y compris l’holoprosencéphalie. La trisomie 18 (syndrome d’Edwards) est généralement due à trois copies distinctes du chromosome 18 et est associée à un mauvais résultat postnatal. Les caractéristiques cliniques comprennent une fente labiale et palatine, une déficience intellectuelle, une incapacité à prospérer, une cardiopathie congénitale, une hypertonie, une micrognathie, un sternum court, des oreilles malformées, des mains serrées, des pieds bas et des ongles hypoplastiques, entre autres. Les trisomies 13 et 18 peuvent être facilement confirmées ou exclues en effectuant une analyse chromosomique (caryotypage).
Les syndromes de microdélétion impliquent généralement la délétion d’une partie d’un chromosome. Ces délétions peuvent être trop petites pour être détectées par caryotypage standard et peuvent nécessiter une technologie FISH (hybridation in situ par fluorescence) ou une technologie de microréseaux pour être détectées. Un syndrome de microdélétion bien connu associé à une fente palatine est le syndrome de délétion 22q11.2 (alias syndrome de Digeorge / Vélocardiofacial). Des anomalies palatines, y compris l’incompétence vélopharyngée, les fentes sous-muqueuses, la luette bifide et la fente palatine, sont observées chez 69% des individus présentant une délétion 22q11.2 et peuvent faire partie de la séquence de Pierre Robin. D’autres résultats cliniques incluent une cardiopathie congénitale, une perte auditive, des caractéristiques dysmorphiques, une déficience immunitaire, une hypocalcémie, des anomalies rénales, des problèmes d’alimentation, des anomalies squelettiques et des troubles psychiatriques. Environ 10% des cas de syndrome de délétion 22q11.2 seraient familiaux. La suppression se sépare de manière autosomique dominante.(6) Le syndrome de Wolf-Hirschhorn, qui est dû à une délétion dans le bras court du chromosome 4, est également associé à des fentes buccales (chez 25% à 50% des individus atteints). Des traits faciaux caractéristiques (y compris une glabelle proéminente conduisant à une “apparence de casque de guerrier grec”), une cardiopathie congénitale, une déficience intellectuelle, des convulsions, une incapacité à prospérer, une micrognathie, des marques ou des fosses préauriculaires et une hypodontie peuvent également être considérés comme faisant partie de la maladie.(7)
Les troubles monogéniques avec fentes orales comprennent le syndrome de Stickler, le syndrome de Treacher Collins et le syndrome de Van der Woude, entre autres. Le syndrome de Stickler est un trouble du collagène avec transmission autosomique dominante et, plus rarement, autosomique récessive. Les caractéristiques communes comprennent une fente palatine (considérée comme faisant partie de la séquence de Pierre Robin ou sans micrognathie), une perte auditive (neurosensorielle et conductrice), des découvertes squelettiques (arthrite précoce, dysplasie spondyloépiphysaire), des anomalies oculaires (myopie élevée, anomalies du vitré) et des traits faciaux caractéristiques (avec sous-développement du maxillaire et du pont nasal, rétrusion de la face médiane). Les tests génétiques pour le syndrome de Stickler peuvent être complexes, car des mutations dans au moins six gènes ont été décrites chez des individus affectés. Environ 90% des patients atteints du syndrome de Stickler présentent des mutations du gène COL2A1 et une forme autosomique dominante de la maladie.(8) Le syndrome de Treacher Collins est une affection autosomique dominante caractérisée par une fente palatine avec ou sans fente labiale chez 28% des personnes atteintes. D’autres anomalies comprennent une hypoplasie des os zygomatiques et de la mandibule, des anomalies de l’oreille externe, un colobome de la paupière inférieure, une perte auditive conductrice, l’absence de cils inférieurs, un déplacement des poils préauriculaires sur les joues et une sténose ou une atrésie choanales. Le diagnostic du syndrome de Treacher Collins est basé sur des résultats cliniques et radiographiques. Des mutations dans au moins trois gènes ont été décrites, avec des mutations de TCOF1 observées chez 78% à 93% des patients.(9) Le syndrome de Van der Woude est caractérisé par la présence de fistules congénitales, généralement bilatérales, paramédiennes de la lèvre inférieure (fosses), ou parfois de petits monticules avec un tractus sinusal provenant d’une glande muqueuse de la lèvre et des fentes buccales (y compris CL / CP et CP). Van der Woude est une affection autosomique dominante associée à des mutations du gène IRF6 (10). L’analyse des conditions d’un seul gène ou de plusieurs gènes nécessite une analyse directe du gène par séquençage et / ou une analyse de délétion / duplication (telle que la MLPA).
Étant donné que les syndromes génétiques avec fente labiale et palatine peuvent être associés à des aneuploïdies, à des microdélétions / microduplications chromosomiques ou à des troubles monogéniques, les tests génétiques peuvent être un processus compliqué. Des antécédents médicaux approfondis, un pedigree de trois générations, des antécédents de grossesse et un examen de dysmorphologie par un généticien clinique peuvent clarifier le tableau clinique et permettre des tests génétiques ciblés. Les technologies plus récentes, y compris les microréseaux, permettront d’identifier les petites microdélétions et microduplications précédemment omises par le caryotypage standard. Malheureusement, cette technique conduit également à l’identification de suppressions et de duplications de signification clinique inconnue, compliquant le processus de conseil génétique. Le dépistage des troubles à gène unique ou des troubles mendéliens nécessite la disponibilité clinique de tests génétiques pour le gène souhaité. Cela peut également être coûteux s’il n’est pas couvert par une assurance médicale. De nouvelles technologies telles que le séquençage de nouvelle génération, le séquençage des exomes ou le séquençage du génome (connu collectivement sous le nom de tests génomiques) sont maintenant disponibles sur le plan clinique. En analysant simultanément des centaines à des milliers de gènes, ces tests augmentent considérablement la puissance et le rendement diagnostiques. Comparés à d’autres techniques, ces tests peuvent apporter une réponse plus rapidement et de manière plus économique. Dans le domaine de la recherche, le séquençage des exomes et du génome a permis d’identifier de nouveaux gènes ainsi que d’élargir les caractéristiques cliniques et le spectre des mutations génétiques. Comme avec la technologie des microréseaux, les tests génomiques peuvent détecter des syndromes sans rapport avec la présentation du patient et / ou la raison du test. Compte tenu des complexités inhérentes aux tests génétiques, le consentement éclairé est nécessaire.
Conclusion
Bien que la fente labiale et palatine soit une anomalie isolée dans la majorité des cas, il existe une forte association entre les fentes buccales et d’autres anomalies et syndromes génétiques. Une évaluation génétique par un généticien clinique et un conseiller en génétique est essentielle pour l’orientation anticipative et pour déterminer les risques de récidive. Les tests génétiques, qui nécessitent un consentement éclairé, peuvent être coordonnés et interprétés lors d’une évaluation génétique.
Anya Revah, MS, est conseillère génétique principale à la Division de génétique médicale de l’Hôpital pour nourrissons et enfants Maimonides à Brooklyn, New York. Elle est également un membre actif du Centre médical Maimonides et de l’équipe multidisciplinaire des Fentes Labiales et palatines de l’Hôpital du comté de Kings. Elle est titulaire d’une maîtrise en Sciences en Conseil génétique de l’Université de Boston à Boston, Massachusetts.
1. Sadler TW. Embryologie médicale de Langman. Neuvième édition. Pages 390-395.
2. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. Pour le Réseau National de prévention des malformations Congénitales. Estimations nationales actualisées de la prévalence à la naissance de certaines malformations congénitales aux États-Unis. 2004-2006. Recherche sur les malformations congénitales (Partie A): Tératologie clinique et moléculaire 2010; 88: 1008-1016.
3. Le Fraser FC. La génétique de la fente labiale et de la fente palatine. Être. J. Hum. Genet. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC, et coll. La consommation périconceptuelle de folate par supplément et prise alimentaire réduit le risque de fente labiale non syndromique avec ou sans fente palatine. Prev Med 2004; 39:689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Perspectives développementales et génétiques sur la séquence de Pierre Robin. Être. J. Med. Genet. 2013; 163C: 295-305.
6. McDonald-McGinn DM, Emanuel BS, Zackai EH. Syndrome de délétion 22q11.2. Sept. 23, 1999. . Dans : Pagon RA, Adam MP, Ardinger HH, et al., éditeur. GénéRalités. Seattle (WA): Université de Washington, Seattle; 1993-2014. Disponible auprès de: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia A, Carey JC, rue South, et al. Syndrome de Wolf-Hirschhorn. Apr. 29, 2002. . Dans : Pagon RA, Adam MP, Ardinger HH, et al., éditeur. GénéRalités. Seattle (WA): Université de Washington, Seattle; 1993-2014. Disponible auprès de : http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Le syndrome d’Ala-Kokko L. Stickler. Jun. 9, 2000. . Dans : Pagon RA, Adam MP, Ardinger HH, et al., éditeur. GénéRalités. Seattle (WA): Université de Washington, Seattle; 1993-2014. Disponible auprès de: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs EW. Syndrome de Treacher Collins. Jul. 20, 2004. . Dans : Pagon RA, Adam MP, Ardinger HH, et al., éditeur. GénéRalités. Seattle (WA): Université de Washington, Seattle; 1993-2014. Disponible à partir de: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte C.B., Saal H.M., Goudy S., et al. Troubles liés à l’IRF6. Oct. 30, 2003. . Dans : Pagon RA, Adam MP, Ardinger HH, et al., éditeur. GénéRalités. Seattle (WA): Université de Washington, Seattle; 1993-2014. Disponible auprès de: http://www.ncbi.nlm.nih.gov/books/NBK1407/.