Les mucopolysaccharidoses: signes diagnostiques précoces chez les nourrissons et les enfants
Les mucopolysaccharidoses (MPS) sont un groupe de maladies cliniquement hétérogènes causées par des carences des enzymes lysosomales nécessaires à la dégradation des glycosaminoglycanes (GAGs).
Les MPS se caractérisent par des manifestations cliniques progressives et systémiques. Malgré leur hétérogénéité biochimique et génétique, différents types partagent des caractéristiques cliniques clés dans différentes combinaisons, y compris la dysplasie articulaire et squelettique avec raideur (sauf la MPS IV où il y a laxité) et douleur, traits faciaux grossiers, opacification de la cornée, hernies inguinales ou abdominales, infections récurrentes des voies respiratoires supérieures, valvulopathie cardiaque, syndrome du canal carpien et atteinte neurologique variable. Ces caractéristiques apparaissent généralement dans les premiers mois de la vie pour les formes sévères et dans la petite enfance pour les formes plus atténuées, mais sont souvent sous-estimées et généralement prises en compte uniquement lorsqu’elles sont clairement évidentes.
La rareté de ces troubles et la variabilité de la présentation clinique conduisent fréquemment à un retard diagnostique; cela peut aller de mois, pour les formes sévères, à des années, pour les formes atténuées (voir Rigoldi et al. dans ce supplément). Néanmoins, en raison de la progression rapide et du besoin urgent d’intervention dans les présentations sévères, même des mois de retard dans le diagnostic peuvent produire des résultats catastrophiques pour la santé future du patient.
Notre objectif est de souligner dans cette revue les signes très précoces des formes sévères qui devraient alerter le pédiatre dès leur première apparition.
- À quels signes / symptômes pouvons-nous nous attendre chez les enfants atteints de formes sévères de MP?
- Quelles sont les différentes présentations des différents types de députés ?
- MPS avec implication somatique et cognitive
- MPS avec une implication principalement neurologique et cognitive
- MPS avec une implication somatique prévalente
À quels signes / symptômes pouvons-nous nous attendre chez les enfants atteints de formes sévères de MP?
Les symptômes et les signes suggérant différentes formes de MPS sont rapportés dans le tableau 1 selon l’âge. Au cours des 6 premiers mois de la vie, des hernies inguinales, des infections respiratoires anormalement fréquentes, des otites et une organomégalie peuvent être observées chez les patients atteints de MPS, en particulier dans le syndrome de MPS I (syndrome de Hurler) qui a l’apparition sévère la plus précoce. De plus, de 6 à 12 mois, ces nourrissons développent très souvent un gibbus (cyphose toraco-lombaire), un souffle cardiaque, une hernie ombilicale, une hypotonie légère et un retard de croissance. Ils peuvent également avoir l’aspect facial grossier typique (Fig. 1). Les MPS II, VI et VII peuvent présenter un phénotype précoce similaire. Les patients atteints de MPS IV ont un phénotype squelettique précoce avec l’apparition d’une dysplasie de la hanche dans les premiers mois de la vie et d’une poitrine de pigeon (pectus carinatum) dans les 12 à 18 premiers mois. Les nourrissons MPS II présentent une implication similaire mais avec une apparition plus tardive. Au cours de la deuxième année de vie, les nourrissons atteints du syndrome de MPS I Hurler développent un retard cognitif tandis qu’un patient MPS II pourrait être identifié uniquement en raison d’une prolifération excessive, d’infections fréquentes des voies respiratoires et de chirurgies multiples; les traits faciaux typiques n’apparaissent que plus tard. Certains patients atteints de MPS II développent également une hyperactivité entre 18 et 24 mois. Au cours de la troisième année de vie, l’hyperactivité et le retard cognitif sont facilement reconnaissables dans MPS II et MPS III.
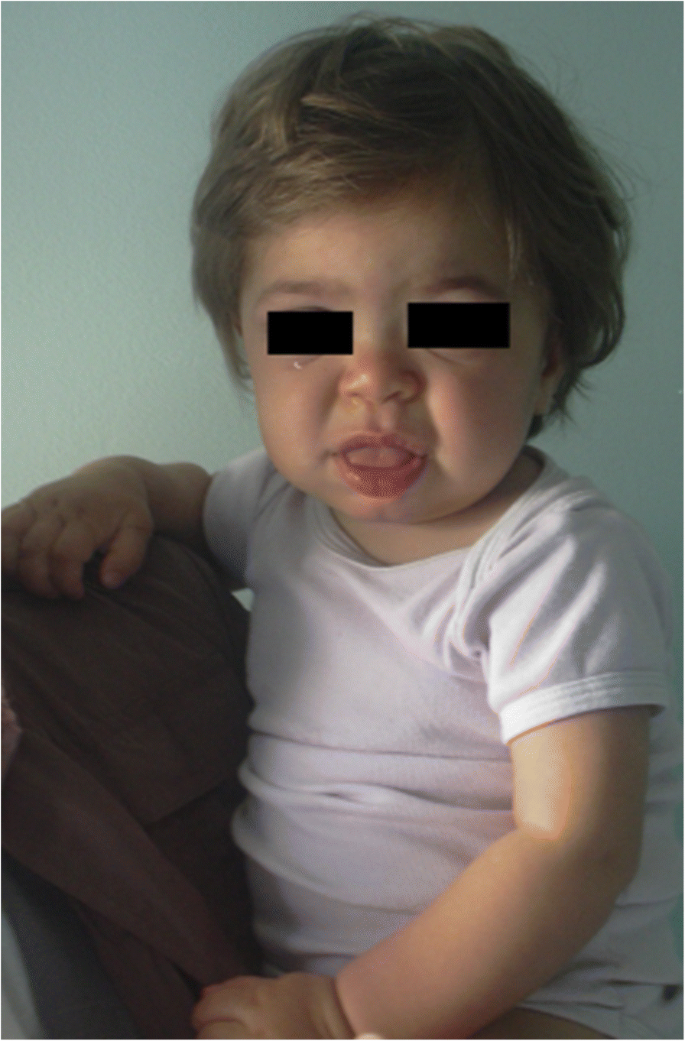
MPS IH chez un patient de 1 an. Faciès grossier : pont nasal plat, macroglossie, bossage frontal
En résumé, les manifestations squelettiques sont typiques et parfois précoces. Le faciès grossier et le retard cognitif ne sont pas des signes saillants précoces.
Quelles sont les différentes présentations des différents types de députés ?
MPS avec implication somatique et cognitive
Mucopolysaccharidose de type I (MPS I; OMIM #252800) (Fig. 1, 2, 3, 4 et 5) est causée par une carence en α-l-iduronidase de l’hydrolase lysosomale, ce qui conduit à une accumulation multisystémique de deux GAGs: le sulfate de dermatane (DS) et le sulfate d’héparane (HS). En fonction de sa gravité, le MPS I est traditionnellement divisé en trois phénotypes cliniques; cependant, ces formes représentent un continuum de gravité de la maladie. Le syndrome de Hurler, la forme la plus grave, se présente généralement au cours de la première année de vie. Les enfants atteints développent rapidement des troubles cognitifs importants et une maladie somatique dans plusieurs systèmes d’organes, entraînant la mort au cours de la première décennie en l’absence de traitement. Les formes atténuées de MPS I, connues sous le nom de syndrome de Hurler–Scheie et de syndrome de Scheie, respectivement, sont caractérisées par l’apparition plus tardive des symptômes, une espérance de vie plus longue et une atteinte légère ou nulle du système nerveux central (SNC). L’hérédité est autosomique récessive et, dans au moins 50% des cas, la prédiction du phénotype est possible sur la base du génotype, tandis que dans les 50% restants, de nouvelles mutations sont détectées. La prévalence est d’environ 1 naissance vivante sur 100 000.

MPS IH. un Gibbus chez un patient de 9 mois. b Radiographie de la colonne vertébrale chez un patient de 3 ans. image par résonance magnétique pondérée c T2 de la colonne vertébrale montrant une cyphose et une sténose du canal vertébral importantes

MPS IH. Preuve radiographique de multiplex de dysostose. un épaississement des côtes chez un patient de 3 ans et une dysplasie de la hanche b chez un autre patient âgé de 18 mois

MPS IH: une main griffe chez un patient de 18 mois; b radiographie de la main.

MPS IH. un épaississement de l’os cortical du crâne et une conformation anormale en “J” de la sella turcica. Dilatation des espaces périventriculaires dans les images de résonance magnétique pondérées b FLAIR et c T-2 d’un patient âgé de 17 mois
Mucopolysaccharidose de type II (MPS II; OMIM #309900) (Fig. 6), également connu sous le nom de syndrome de Hunter, est le résultat d’une déficience de l’enzyme iduronate-2-sulfatase (I2S) avec accumulation conséquente de DS et de HS. La prévalence est d’environ 1 sur 140 000 à 156 000 naissances vivantes masculines. Il s’agit notamment du seul MPS lié à l’X, et les porteuses sont généralement asymptomatiques; cependant, elles peuvent être exceptionnellement affectées en raison d’anomalies du chromosome X, de l’homozygotie ou de l’inactivation asymétrique de l’X. L’expression phénotypique couvre également un large spectre de gravité clinique. Dans les cas graves, une atteinte neurologique progressive marquée et une maladie somatique coexistent, et la mort survient généralement au cours de la deuxième décennie de la vie. D’autres patients présentant un dysfonctionnement neurologique minimal ou nul peuvent présenter une atteinte somatique sévère tandis que, à l’extrémité opposée du spectre, il y a des patients avec seulement des manifestations somatiques minimales et une intelligence normale qui survivent à l’âge adulte. Les signes et symptômes précoces sont partagés par les formes sévères de MPS I et II.

MPS II. Un patient de 3 ans avec seulement de légères caractéristiques faciales
Des cas sporadiques d’hydrops fetalis sont décrits dans MPS I, bien qu’en général, ces enfants semblent normaux à la naissance. Les premiers signes de MPS I apparaissent généralement au cours des tout premiers mois de la vie, tandis que ceux de MPS II surviennent généralement un peu plus tard. Kiely et coll. , dans une étude rétrospective chez 55 patients Hurler, a observé que des symptômes respiratoires, des difficultés d’alimentation, une hernie inguinale et une otite moyenne étaient apparus à un âge médian ≤ 6 mois, et une cyphose, des anomalies cardiaques, un tour de tête élargi, une hypotonie, une organomégalie, une opacification cornéenne, une restriction articulaire et une hernie ombilicale sont apparus entre 6 et 12 mois. La hernie ombilicale est l’une des premières caractéristiques de présentation des MPS I et II, et des hernies inguinales sont rapportées chez environ 60% des patients.
Un autre indice initial courant pour les MPS I et II peut être des infections récurrentes des voies respiratoires supérieures avec une augmentation des sécrétions. Les otites fréquentes, les rhinites chroniques récurrentes et les écoulements nasaux persistants sans infection évidente ne sont pas spécifiques, mais peuvent aider à renforcer la suspicion diagnostique si elles sont associées à des signes plus spécifiques. Le stockage du BÂILLON dans l’oro-pharynx entraîne une hypertrophie des amygdales et des végétations adénoïdes et contribue aux complications des voies respiratoires supérieures. Une autre découverte fréquente est la respiration bruyante, en particulier la nuit, associée aux stades ultérieurs à une apnée obstructive du sommeil. En raison d’infections fréquentes de l’oreille moyenne et d’une dysostose des osselets de l’oreille moyenne, la perte auditive est fréquente, avec des déficits conducteurs et neurosensoriels. La trachéobronchomalacie est couramment observée et peut entraîner une obstruction aiguë des voies respiratoires ou un collapsus, ce qui est l’une des principales causes de décès au cours des années suivantes.
L’adénoïdectomie, l’amygdalectomie et la tympanostomie, ainsi que la réparation d’une hernie, sont les interventions chirurgicales les plus fréquentes et précoces chez les patients atteints de MPS I et II, souvent effectuées avant de connaître le diagnostic (dans plus de 50% des cas).
La diarrhée récurrente est assez fréquente aux stades précoces de la MPS I et peut également être présente chez les patients atteints de MPS II présentant une atteinte neurologique, alors qu’elle est rare chez les patients légèrement atteints.
Déformation du Gibbus (cyphose dorso-lombaire) (Fig. 2) devient cliniquement apparent à un âge médian de 8 ans.6 mois à 1 an chez MPS I, représentant un marqueur assez spécifique et précoce des MPS en général. Les corps vertébraux semblent aplatis et becqués, ce qui peut entraîner des complications ultérieures telles que le piégeage du nerf rachidien, une lésion aiguë de la colonne vertébrale et une instabilité atlanto-occipitale; il faut donc prendre des précautions particulières pour éviter la luxation de l’articulation atlanto-axiale pendant l’anesthésie générale et la chirurgie. Après l’âge de 1 an, les contractures articulaires et le genu valgum représentent d’autres signes fréquents de déficience articulaire et de dysplasie squelettique progressive, qui sont facilement détectables à l’âge de 2 ans chez tous les nourrissons atteints de MPS. Toutes ces manifestations squelettiques ensemble, typiques de la MPS, sont appelées multiplex de dysostose. L’épaississement des côtes peut être vu sur les radiographies (Fig. 3a), les os longs sont courts avec de larges tiges et les genoux sont sujets aux déformations du valgus et du varus. La dysostose phalangienne et l’épaississement synovial entraînent une déformation caractéristique de la main de la griffe (Fig. 4), ce qui est une autre caractéristique typique qui peut susciter la suspicion de la maladie. La dysplasie de la hanche est souvent la raison pour laquelle ces enfants peuvent être vus pour la première fois par des orthopédistes. En règle générale, le bassin est mal formé, les têtes fémorales sont petites et la coxa valga est commune, avec une déformation progressive et débilitante de la hanche (Fig. 3b).
Il existe une différence notable entre les MPS I et les MPS II; chez les MPS I, la croissance squelettique ralentit à l’âge de 3 ans, tandis que les patients atteints de MPS II présentent une prolifération au cours des 5-6 premières années, ralentissant leur croissance par la suite.
Le grossissement des traits du visage (nez large avec narines évasées, crêtes supraorbitales proéminentes, grandes joues arrondies, lèvres épaisses, langue saillante élargie) causé par le stockage de GAGs dans les tissus mous de la région orofaciale et des os du visage devient apparent au cours des 2 premières années, à un âge médian de 1,1 an pour la maladie de Hurler et entre 18 mois et 4 ans pour la forme sévère de la maladie de Hunter, bien que de subtils changements phénotypiques puissent être détectés plus tôt (dès la seconde moitié du première année) par des pédiatres ayant une expérience spécifique (Fig. 1). La macrocéphalie devient progressivement plus évidente et la scaphocéfalie est fréquente (Fig. 5), mais la microcéphalie est également possible chez les patients Hurleurs. L’hirsutisme facial et corporel est toujours évident à l’âge de 24 mois. La peau peut être épaissie et inélastique. Notamment, certains patients atteints de MPS II développent une lésion cutanée distinctive, décrite comme des papules blanc ivoire (en forme de caillou) sur le haut du dos et les côtés des bras, pathognomonique du syndrome de Hunter.
La cardiomyopathie et l’épaississement et le raidissement progressifs des folioles valvulaires chez les patients atteints de MPS I peuvent entraîner une régurgitation mitrale et, moins fréquemment, une régurgitation aortique. Un murmure peut être détecté dans les premiers mois de la vie et a parfois été signalé comme le premier signe de la maladie. L’atteinte cardiaque est également présente chez presque tous les patients atteints de MPS II, mais cela commence vers l’âge de 5 ans. Une maladie valvulaire peut entraîner une hypertrophie ventriculaire droite et gauche et une insuffisance cardiaque.
La protubérance de l’abdomen causée par une hépatosplénomégalie progressive est facilement évidente après l’âge de 6 mois, bien que le stockage des GAGs dans le foie et la rate n’entraîne pas de dysfonctionnement des organes.
Une opacification cornéenne survient chez presque toutes les personnes atteintes de MPS I avant l’âge de 15 mois, ce qui peut entraîner une déficience visuelle grave. Au contraire, l’opacification cornéenne n’est pas une caractéristique typique du syndrome de Hunter, mais l’examen à la lampe à fente peut révéler des lésions cornéennes discrètes qui n’affectent pas la vision. Un dysfonctionnement de la rétine peut être détecté, tandis que le glaucome n’est pas une découverte courante.
Le développement psychomoteur précoce est généralement détecté comme normal, mais chez MPS I, le retard de développement est généralement évident à l’âge de 18 mois, avec un déclin mental progressif conduisant à une déficience intellectuelle grave. Les compétences linguistiques sont très limitées chez ces enfants. Un retard global des étapes de développement dans MPS II devient évident à partir de 2 ans. Cependant, la principale caractéristique neurologique de la maladie de Hunter sévère est représentée par des troubles du comportement marqués, tels que l’hyperactivité, l’obstination et l’agressivité, qui ne sont généralement pas observés chez les personnes dont le phénotype est atténué.
La mucopolysaccharidose de type VII (MPS VII; OMIM # 253220), également appelée syndrome de Sly, est une MPS ultra-rare caractérisée par une activité déficiente de la β-glucuronidase, avec stockage lysosomal du sulfate de chondroïtine (CS), DS et HS, entraînant un dysfonctionnement cellulaire et organique. Le premier patient a été décrit par Sly et al. en 1973 et un total de 145 patients ont été signalés dans la littérature jusqu’à présent.
La présentation clinique et la progression de la maladie de MPS VII couvrent un large spectre de gravité, allant des manifestations précoces, sévères et multisystémiques et de la mort dans les tout premiers mois, à un phénotype plus doux avec apparition plus tardive, intelligence normale ou quasi normale et survie plus longue.
Les caractéristiques phénotypiques des patients atteints de MPS VII ressemblent à celles des MPS I et MPS II (petite taille, dysplasie squelettique, macrocéphalie, hypertrophie gingivale, otites récurrentes, hépatosplénomégalie, hernies, atteinte cardiaque, diminution de la fonction pulmonaire et troubles cognitifs). Une proportion étonnamment élevée de patients décrits (41%) ont des antécédents d’hydrops fœtaux néonataux non immunisés (NIHF). Malgré l’apparition prénatale de la maladie chez ces patients, 13 sur 23 ont survécu à la petite enfance avec une évolution légère à intermédiaire jusqu’à la fin de l’adolescence. Ainsi, la présence de NIHF ne permet pas, à elle seule, de prédire la gravité possible de l’évolution clinique si le patient survit à la petite enfance. Un autre signe précoce est la macrocranie, tandis que l’atteinte des valves cardiaques et les infections répétées des voies respiratoires supérieures et inférieures apparaissent au cours des 2 premières années de la vie. Un retard mental modéré et une perte auditive progressive avec troubles de la parole sont également évidents avec le temps.
En résumé, les formes sévères de MPS I ont un début multisystémique très précoce (dans les 6 mois de la vie); La MPS II est similaire mais avec un début plus tardif, et la MPS VII a un début prénatal avec une NIHF fréquente et une atteinte sévère précoce des voies respiratoires supérieures et inférieures.
MPS avec une implication principalement neurologique et cognitive
Mucopolysaccharidoses de type III (MPS III, également connu sous le nom de syndrome de Sanfilippo; Fig. 7) a une présentation neurologique répandue. Le MPS III est un trouble du stockage lysosomal causé par une déficience de l’une des quatre enzymes impliquées dans le catabolisme de l’HS. Quatre sous—types différents sont connus — MPS III de type A (OMIM # 252900), de type B (OMIM # 252920), de type C (OMIM # 252930) et de type D (OMIM # 252940) – chacun en raison d’un déficit enzymatique différent: type A, gène sulfamidase / SGSH; type B, gène alfa-N-acétylglucosaminidase / NAGLU; type C, alfa-glucosaminide N-acétyltransférase /NAGLU Gène HGSNAT; type D, gène N-acétilglucosamina-6-solfato sulfatasi/GNS). Les quatre types (A à D) ont une hérédité autosomique récessive. La MPS III est la plus fréquente des MPS avec une prévalence estimée entre 0,3 et 4.1 cas pour 100 000 nouveau-nés, selon le sous-type et la race.

MPS II A. Le même patient à a 2,5 et b 5 ans. Notez l’expression différente du visage, montrant la progression de l’atteinte neurologique
Les patients atteints du syndrome de Sanfilippo présentent une déficience cognitive progressive au cours des deuxième et troisième années de vie. Contrairement à la majorité des députés, les caractéristiques somatiques sont relativement moins évidentes et l’implication du SNC est visible. Le retard de développement de la parole est généralement le premier signe remarqué par les parents mais les principales préoccupations peuvent être des problèmes de comportement (hyperactivité, comportement anxieux et agressif, troubles du sommeil), suivis d’un déclin mental progressif. Ces symptômes peuvent entraîner un diagnostic erroné comme troubles du comportement, trouble déficitaire de l’attention avec hyperactivité (TDAH) et troubles du spectre autistique. L’épilepsie peut également être présente.
Les troubles du sommeil, dont l’incidence est rapportée jusqu’à 80-90%, sont une caractéristique commune. Ils consistent en des difficultés d’endormissement et un réveil nocturne fréquent, avec inversion complète du rythme jour–nuit chez certains patients.
Toutes ces manifestations sont très difficiles à gérer et ont un impact psychologique énorme sur la qualité de vie de toute la famille.
En général, bien que le phénotype puisse être très similaire dans les quatre sous-types, l’évolution clinique chez Sanfilippo B et C semble être caractérisée par un phénotype moins sévère.
Les symptômes non neurologiques sont généralement moins prononcés chez MPS III que chez les autres MPS. Des infections récurrentes de l’oreille, du nez et de la gorge sont observées à un plus jeune âge. Une otite récurrente, des défauts d’osselets de l’oreille moyenne et des anomalies de l’oreille interne peuvent provoquer une surdité. Un excès de sécrétions épaisses et des modifications anatomiques peuvent provoquer une obstruction des voies respiratoires. Dans les premières années de la vie, la diarrhée est fréquemment rapportée, alors que la constipation est plus fréquente chez les patients plus âgés.
L’examen physique peut montrer des traits faciaux grossiers, bien que ceux-ci soient moins prononcés. Les patients ont une macrocéphalie, de larges sourcils avec un évasement médial et des synophrys, les cheveux sont généralement secs et grossiers et l’hypertrichose est fréquente. Chez les patients plus jeunes, une hépatomégalie légère et des hernies ombilicales et inguinales sont fréquemment observées. Les contractures rares et souvent légères se trouvent principalement dans les coudes. La croissance est affectée chez environ la moitié des patients âgés de plus de 12 ans.
En résumé, la MPS III ou maladie de Sanfilippo présente une atteinte neurologique importante avec des signes somatiques très légers. Les tout-petits âgés de 2 à 3 ans développant une hyperactivité, un TDAH et un comportement agressif peuvent être soupçonnés d’avoir des MPS III.
MPS avec une implication somatique prévalente
MPS IV et MPS VI présentent uniquement une implication somatique. Ce sont des conditions progressives qui épargnent les capacités intellectuelles et affectent principalement le squelette (MPS IVA), ou tous les organes / systèmes du corps (MPS VI). L’héritage est autosomique récessif. Le taux d’aggravation des symptômes varie selon les personnes touchées.
Mucopolysaccharidose IV (MPS IV, également connu sous le nom de syndrome de Morquio; Fig. 8) se présente comme deux troubles génétiquement distincts, chacun avec un déficit enzymatique différent: MPS IVA (syndrome de Morquio A; OMIM # 253000) dû à une carence en N-acétylgalactosamine-6-sulfatase (gène GALNS), entraînant une accumulation de kératan-sulfate (KS) et de CS; et MPS IVB (syndrome de Morquio B; OMIM # 253010) dû à une déficience de l’activité bêta-galactosidase conduisant à une excrétion urinaire de KS et d’oligosaccharides comme chez les patients GM1 . MPS IVB est en effet allélique aux différentes formes de gangliosidose GM1, mais n’a pas la détérioration psychomotrice observée dans la gangliosidose GM1.

MPS IVA. un patient de 4 ans avec des radiographies b antéro-postérieures et c latérales montrant des côtes “en forme de pagaie” et des vertèbres aplaties et arrondies avec un aspect typique de “bec antérieur
Les patients atteints de MPS IVA sévère sans activité enzymatique détectable révèlent leurs premiers symptômes généralement au cours de la première année de vie, bien qu’ils soient rarement diagnostiqués avant l’âge de 4 à 5 ans. Les informations sur l’histoire naturelle des patients atteints de Morquio A proviennent principalement de deux collections de données très variées. Le taux de croissance réduit commence à environ 18 mois et la croissance s’arrête à environ 7 ou 8 ans. Contrairement aux autres MPS, les articulations de Morquio A sont généralement laxistes et très flexibles (hypermobiles), mais certaines articulations (généralement les hanches et les épaules) peuvent avoir une amplitude de mouvement limitée. Les patients présentant une telle atteinte squelettique peuvent recevoir un diagnostic ou subir une évaluation de la dysplasie spondyloépiphysaire, de la pseudoachondroplasie, de la dysplasie épiphysaire multiple ou de la maladie bilatérale de Legg-Calvé-Perthes.
Une caractéristique constante est l’hypoplasie odontoïde; ainsi, une luxation de l’articulation atlanto-occipitale peut se produire entraînant une compression et des lésions de la moelle bulbaire et de la moelle épinière, entraînant une paralysie ou même la mort si la stabilisation cervicale n’est pas réalisée précocement.
Les patients MPS IVB ont une manifestation clinique très similaire mais avec une petite taille moins proéminente. Ils ont des traits du visage légèrement grossiers, une perte auditive, des opacités cornéennes, une cardiopathie valvulaire et des infections fréquentes des voies respiratoires supérieures. Ils peuvent également présenter une hernie inguinale et une hépatomégalie légère. Les caractéristiques dysplasiques squelettiques comprennent la platyspondylie, l’hypoplasie odontoïde avec subluxation cervicale possible, la cypho-scoliose, la coxa valga et les ailes iliaques rétrécies.
Mucopolysaccharidose VI (MPS VI, également connu sous le nom de syndrome de Maroteaux-Lamy; OMIM #253200; Fig. 9) est due à des mutations pathogènes du gène de l’arylsulfatase B (ARSB) situé sur le chromosome 5. En conséquence, l’activité réduite ou absente de l’enzyme arylsulfatase B (ASB) (N-acétylgalactosamine 4-sulfatase) altère la dégradation de la DS déterminant son accumulation cellulaire.

MPS VI. Un garçon de 2 ans avec un visage grossier évident et une dysostose squelettique
Les patients sans activité enzymatique détectable présentent la forme sévère de MPS VI et présentent des symptômes dans la petite enfance. Leur phénotype somatique est similaire au syndrome de Hurler. Dans la plupart des cas, à l’âge de 2 ou 3 ans, des lésions squelettiques graves et progressives, appelées multiplex de dysostose, deviennent évidentes. Cette dysplasie squelettique comprend une dysplasie de la hanche avec une tête fémorale dysplasique, des corps vertébraux anormaux, des clavicules irrégulières, un cubitus et un radius distaux hypoplasiques et des os métacarpiens dysplasiques et courts; les os du carpe et du tarse sont hypoplasiques et ont un profil irrégulier. La vitesse de croissance ralentit souvent après la première année de vie, avec une hauteur finale généralement inférieure à 120 cm. Comme chez les autres députés, un signe précoce est les traits faciaux grossiers. De plus, les patients présentent un abdomen saillant typique, une hépatomégalie, une hernie ombilicale et / ou inguinale et une atteinte cardiaque au début de la petite enfance. Bien qu’aucune déficience intellectuelle primaire ne soit présente, une hydrocéphalie peut être observée chez certains patients présentant une hypertension intracrânienne et un œdème papillaire conséquents. Le pectus carinatum, les articulations raides et contractées, la scoliose et la cyphose et la compression de la moelle épinière cervicale s’aggravent avec le temps.
En résumé, les MPS IV et VI ne montrent pas d’implication du SNC. Le MPS IV est un MPS unique en montrant un laxisme articulaire, tandis que l’apparition sévère du MPS VI est similaire à celle observée chez le MPS I.