Applicazione di test di complementazione di frammenti di proteine in biologia cellulare
- Introduzione
- Principio
- Limitazioni
- Controlli standard per uno studio PCA
- Applicazione di PCA nella progettazione di proteine: Library vs. Library Screening per proteine che interagiscono in modo ottimale
- Applicazione della PCA allo screening della libreria cDNA nelle cellule di mammifero
- Usando PCA come righello molecolare: Studi sui recettori
- Mappatura delle reti biochimiche
- Conclusione
- Riconoscimenti
Introduzione
I percorsi biochimici sono in realtà sistemi di assemblaggio e smontaggio dinamico di complessi proteici, e quindi, gran parte della moderna ricerca biologica si occupa di come, quando e dove le proteine interagiscono con altre proteine coinvolte nei processi biochimici. La domanda di approcci semplici per studiare le interazioni proteina-proteina, in particolare su larga scala, è cresciuta di recente con i progressi nei progetti sul genoma, poiché l’associazione di sconosciuti con prodotti genici noti fornisce un modo cruciale per stabilire la funzione di un gene. È stato con questa sfida in mente che il nostro laboratorio ha sviluppato test di complementazione di frammenti di proteine (PCAS). In questa strategia, due proteine di interesse (proteine A e B) sono fuse a frammenti complementari di una proteina reporter (un enzima, una proteina fluorescente, ecc.). Se le proteine A e B interagiscono, i frammenti del reporter vengono riuniti, piegati nella struttura nativa del reporter e ricostituiscono la sua attività (Figura 1). Le proteine reporter PCA sono state scelte come quelle che producono una varietà di attività rilevabili, tra cui segnali fluorescenti, luminescenti e colorimetrici, nonché semplici saggi di selezione della sopravvivenza (1-14). Abbiamo dimostrato che la strategia PCA ha le seguenti capacità: (i) consente il rilevamento di interazioni proteina-proteina in vivo e in vitro in qualsiasi tipo di cellula; (ii) consente il rilevamento delle interazioni proteina-proteina in appositi compartimenti subcellulari o organelli; (iii) consente il rilevamento di interazioni che sono specificamente indotta in risposta a sviluppo, nutrizionali, ambientali, o ormone-indotta segnali; (iv) che permette il monitoraggio di cinetica ed equilibrio di assemblaggio di proteine in cellule; e (v) permette di screening per il romanzo di interazioni proteina-proteina in qualsiasi tipo di cellula (2,3,6,9)(15-19).
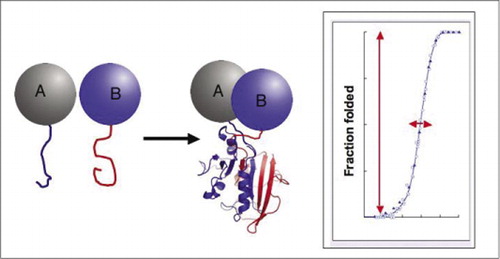
Se le due proteine interagiscono, i frammenti reporter vengono riuniti, piegati nella struttura nativa della proteina reporter e la sua attività viene ricostituita (a sinistra). Questi test di complementazione proteina-frammento (PCAs) hanno caratteristiche fisiche che li rendono particolarmente utili come reporter di complessi proteici dinamici. Sulla destra c’è una curva di piegatura della proteina in cui l’asse x è un parametro variabile (ad es., concentrazione di un frammento rispetto ad un altro). L’elevata cooperatività di questo processo (aumento estremamente forte della frazione di specie piegate su un intervallo molto ristretto) significa che i saggi hanno un’enorme gamma dinamica, rendendo il rilevamento di un complesso un fenomeno virtuale tutto o nessuno. Ciò contrasta con metodi come il trasferimento di energia di risonanza a fluorescenza (FRET), che ha una gamma dinamica molto bassa e richiede un’attenta ottimizzazione di un certo numero di parametri. Al contrario, misurare la formazione di complessi proteici mediante PCA non è più difficile della misurazione dell’attività dell’enzima reporter intatto.
Principio
Abbiamo dimostrato il principio della PCA a partire dall’enzima diidrofolato reduttasi (DHFR) come reporter (1). Era ovvio che se il ripiegamento dell’enzima dai suoi frammenti (come rilevato dalla ricostituzione dell’attività) era assolutamente dipendente dal legame tra le proteine interagenti, allora il sistema descritto è, infatti, un rivelatore delle interazioni. Noi e altri da allora abbiamo dimostrato che questo principio può essere generalizzato a un certo numero di enzimi tra cui Gaussia e Renilla luciferasi, TEM β-lattamasi, così come la proteina fluorescente verde (GFP) e le sue varianti (1-14). Una caratteristica cruciale dei frammenti di PCA è che sono progettati per non piegarsi spontaneamente senza essere portati in prossimità dall’interazione delle proteine a cui sono fusi (1,20). Se si verificasse una piegatura spontanea, la PCA semplicemente non funzionerebbe. La piegatura spontanea porterebbe a un falso segnale positivo, una situazione che confonderebbe irrimediabilmente l’interpretazione degli schermi della libreria in vivo (prevista per essere un’applicazione importante). A differenza di PCA, esistono sistemi di analisi basati su β-galattosidasi e split inteins che assomigliano a PCA, ma che sono concettualmente e praticamente diversi (21,22). In entrambi i casi, le ben note subunità naturali e che associano spontaneamente gli enzimi sono fuse alle proteine interagenti. Il problema centrale qui è che le subunità, anche se debolmente associate, sono sempre in grado di farlo in una certa misura, il che significa che c’è uno sfondo costante di assemblaggio spontaneo.
Limitazioni
La strategia PCA è generale, nel senso che non è limitata a un singolo reporter enzimatico, ed è stata concepita in diverse forme, ognuna delle quali è più adatta per affrontare una domanda specifica. Per esempio, semplici PCAS sopravvivenza-selezione, come quelli basati su DHFR, sono più utili per la selezione biblioteca, mentre luminescenza o fluorescenza lettura PCAs sono i migliori per gli studi della dinamica spaziale e temporale dei complessi proteici. Poiché le proteine di fusione possono essere espresse in cellule che sono modelli rilevanti per lo studio di uno specifico percorso biochimico, sono probabilmente nel loro stato biologico nativo comprese le corrette modifiche post-traduttive (ovviamente i frammenti di PCA stessi non devono interferire con il targeting o la modifica delle proteine, e questo deve essere testato).
Tra i PCAS più semplici e quindi più popolari ci sono quelli basati su proteine fluorescenti (come GFP e varianti), perché il segnale è fornito dal fluoroforo intrinseco(7-9) (14,15,17,23). Tuttavia, le proteine fluorescenti devono essere espresse ad alti livelli per assicurare che il segnale sia al di sopra della fluorescenza cellulare di fondo, e le proteine fluorescenti PCA sono state dimostrate irreversibili, il che può essere utile (intrappolando e visualizzando complessi rari) ma potrebbe anche portare a un’interpretazione errata del turnover o della localizzazione delle proteine interagenti (8,23,24). D’altra parte, i PCAS basati su DHFR e β-lattamasi come reporter hanno dimostrato, sulla base di prove indirette, di essere reversibili in seguito all’interruzione delle interazioni, mentre un PCA basato su Gaussia luciferasi è stato dimostrato direttamente reversibile (2,3,6). La reversibilità della PCA consente quindi di rilevare gli aspetti cinetici e di equilibrio dell’assemblaggio e dello smontaggio del complesso proteico nelle cellule viventi (Figura 2).
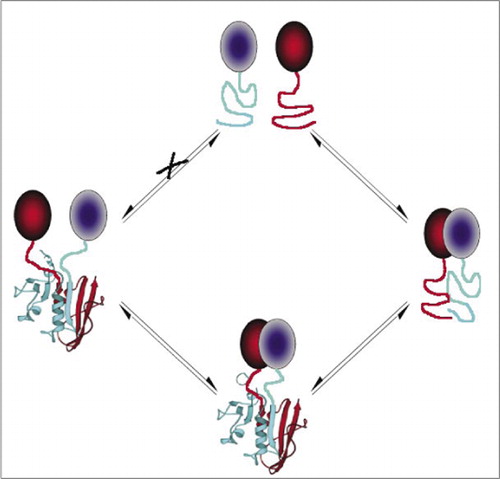
Ciò impedisce l’associazione spontanea dei frammenti (via X) che può portare a un falso segnale. Allo stesso modo, vengono selezionati frammenti per i quali si dovrebbe verificare lo svolgimento spontaneo dei frammenti quando il complesso proteico viene interrotto (lato sinistro).
Controlli standard per uno studio PCA
La strategia PCA richiede che i frammenti della proteina reporter si assemblino e si pieghino dopo che le proteine di interesse hanno formato un complesso. L’assemblaggio e la corretta piegatura del reporter dipendono dal recupero sia della geometria strutturale intrinseca alle proteine reporter che del complesso formato dalle proteine interagenti. Questa è una delle principali distinzioni dei saggi PCA rispetto ai saggi di trasferimento di energia di risonanza a fluorescenza (FRET) o di trasferimento di energia di risonanza a bioluminescenza (BRET) o di lievito a due ibridi, e questa caratteristica ci ha permesso di eseguire uno studio basato sulla struttura del recettore dell’eritropoietina (19). In genere inseriamo un linker polipeptidico flessibile a 10 aminoacidi costituito da (Gly.Gly.Gly.Gly.Ser) 2 tra la proteina di interesse e il frammento reporter PCA (per entrambe le fusioni). Questo linker è stato scelto perché è il più flessibile possibile, e abbiamo osservato empiricamente che i linker di questa lunghezza sono sufficientemente lunghi da consentire ai frammenti di trovare l’un l’altro e piegarsi, indipendentemente dalla dimensione delle proteine interagenti a cui i frammenti sono fusi (16).
Per garantire che non si verifichino risposte non specifiche, è necessario eseguire una serie di controlli. Questi controlli potrebbero includere quanto segue, sebbene il primo sia il più importante: (i) proteine non interagenti. Non si deve osservare una risposta PCA se si utilizzano proteine non interagenti come partner PCA; né la sovraespressione di una proteina non interagente da sola dovrebbe competere per l’interazione nota. (ii) Mutazioni dell’interfaccia proteica partner. Un punto o mutazione delezione di un partner che è noto per interrompere un’interazione dovrebbe anche impedire una risposta PCA. – Concorrenza. Una risposta di PCA dovrebbe essere diminuita dalla sovraespressione simultanea di una o dell’altra delle proteine interagenti che non è fusa ad un frammento complementare di PCA. iv) Scambio di frammenti. Un’interazione osservata fra due proteine dovrebbe accadere anche se le proteine sono scambiate con i rispettivi frammenti del reporter.
Applicazione di PCA nella progettazione di proteine: Library vs. Library Screening per proteine che interagiscono in modo ottimale
Tra le prime applicazioni di un PCA è stato un problema di progettazione di proteine. Il test DHFR PCA è stato utilizzato in Escherichia coli per vagliare due librerie di sequenze di leucina zipper-forming progettate in modo complementare con 1010 potenziali coppie interagenti, di cui potremmo praticamente coprire 106. Abbiamo dimostrato che lo schermo PCA selezionato sia per la specificità di legame ottimale, sia per la solubilità e l’espressione delle cerniere interagenti (18,25). La caratteristica più importante di questo approccio è che è stato possibile schermare contemporaneamente due librerie l’una contro l’altra, un processo non facilmente raggiungibile con schermi lievito due ibridi comparabili. La semplicità di questo approccio e la natura specifica delle informazioni ottenute sulla strategia di progettazione suggeriscono un’ampia utilità del DHFR PCA nella progettazione di proteine e negli esperimenti di evoluzione diretta. Mostra anche che PCA integra le strategie di visualizzazione dei fagi, poiché l’intera selezione, ottimizzazione e test di stringenza vengono eseguiti in vivo, rendendo questo approccio facilmente eseguibile.
Applicazione della PCA allo screening della libreria cDNA nelle cellule di mammifero
Un primo passo nella definizione della funzione di un nuovo prodotto genico consiste nel determinare le sue interazioni con altri prodotti genici. Tuttavia, un approccio di screening basato sull’interazione puramente proteica (come il lievito a due ibridi) è limitato, perché ti dice solo che due proteine interagiscono, senza fornire altre informazioni che potrebbero collegare una proteina alla sua funzione. Pertanto, abbiamo dimostrato che la PCA può essere utilizzata in una strategia di screening della libreria cDNA che combina un semplice schermo di interazione proteica basato sulle cellule con test funzionali specifici che forniscono una convalida iniziale della rilevanza biologica dell’interazione (9). Il primo passo consiste nello screening delle interazioni fisiche tra l’esca e una libreria di proteine prede codificate cDNA, monitorando la ricostituzione del reporter PCA in cellule viventi intatte. Una caratteristica importante di questo primo passo è che le interazioni possono essere rilevate direttamente e tra proteine a lunghezza intera in cellule in cui la proteina esca normalmente funziona, assicurando così che il targeting subcellulare necessario, le modifiche post-traduttive e le interazioni con altre proteine possono verificarsi. Ovviamente, per la validità sperimentale, i frammenti di PCA devono essere dimostrati di non interferire con il targeting o la modifica delle proteine. Nella seconda fase, l’interazione proteica può essere convalidata funzionalmente, come segue: in primo luogo, l’interazione proteica, rilevata da PCA, deve essere perturbata da agenti, come ormoni o inibitori specifici, che sono noti per modulare la specifica via biochimica in cui le proteine partecipano. Lo abbiamo dimostrato per il DHFR PCA e utilizzato questa proprietà per mappare le vie di segnalazione nelle cellule viventi di mammiferi (16). In secondo luogo, la localizzazione subcellulare dell’interazione proteica, nuovamente rilevata da PCA, potrebbe essere alterata da agenti che modulano la via. Pertanto, la strategia di screening basata su PCA combina una semplice fase di screening con saggi funzionali diretti. Noi e altri abbiamo applicato questa strategia per l’identificazione di nuovi substrati o regolatori della serina/treonina protein chinasi, PKB/Akt (9,15,26,27).
Usando PCA come righello molecolare: Studi sui recettori
Una caratteristica speciale delle strategie PCA è che, se conosciamo la struttura tridimensionale dell’enzima reporter, è possibile prevedere con precisione quanto devono essere vicini i frammenti per assicurare che l’enzima si pieghi correttamente e abbia un’attività misurabile. Questo fatto è stato messo al lavoro per testare un modello allosterico strutturale per l’attivazione del recettore dell’eritropoietina dimerica (EpoR) utilizzando il DHFR PCA, e l’approccio potrebbe essere esteso allo studio delle transizioni allosteriche nelle interfacce proteiche dimeriche o multimeriche (19). Nel caso di EpoR, i domini transmembrana del dimero del recettore sono stati mostrati separati da 73 Å, come osservato nella struttura cristallina di EpoR non legato. È stato ragionato che se questo stato inattivo esistesse sulla membrana di una cellula vivente, allora i frammenti di DHFR fusi ai termini C dei domini transmembrana si piegherebbero solo se un ligando inducesse un cambiamento di conformazione che permettesse ai frammenti di avvicinarsi abbastanza insieme da assicurare che la precisa struttura tridimensionale di DHFR potesse essere formata (19,28). Ciò richiederebbe che la N termini dei frammenti sia 8 Å a parte. L’inserimento di peptidi linker flessibili tra il dominio transmembrana e i frammenti di DHFR ci ha permesso di sondare la distanza tra i punti di inserimento del dimero del dominio extracellulare e confermare che i linker abbastanza lunghi da estendersi a 73 Å erano necessari per il DHFR per piegarsi dai suoi frammenti.
Mappatura delle reti biochimiche
I macchinari biochimici cellulari per il metabolismo, le cascate di segnalazione e il ciclo cellulare sono esempi di assemblaggio e smontaggio dinamico di complessi macromolecolari. Questi sono definiti raggruppando proteine interagenti in base alle loro risposte simili a un insieme di perturbazioni (ormoni, metaboliti, inibitori enzimatici, ecc.). Le interazioni proteina-proteina possono essere utilizzate per collegare una proteina di funzione sconosciuta a proteine che sono note per essere coinvolte in un processo biochimico noto. Abbiamo dimostrato che il profiling farmacologico (monitoraggio degli effetti di farmaci specifici per via e ormoni proteici sulle interazioni proteina-proteina) e la determinazione della posizione cellulare delle interazioni proteina-proteina possono essere raggiunti utilizzando PCAs (9)(15-17)(26). L’analisi di questi risultati consente una rappresentazione di come le reti biochimiche si evolvono nel tempo e nello spazio e in risposta a stimoli specifici. Come prova di principio, abbiamo riportato l’applicazione di questa strategia alla mappatura di una via di trasduzione del segnale mediata dalle tirosin chinasi del recettore (RTK) (16). I profili farmacologici e la posizione cellulare delle interazioni che abbiamo osservato ci hanno permesso di posizionare ciascun prodotto genico nel suo punto rilevante nei percorsi (Figura 3). Dai risultati della nostra analisi, è emersa una mappa dell’organizzazione della rete RTK coerente con i modelli esistenti, ma che includeva anche diverse nuove interazioni. La capacità di monitorare una rete di interazioni proteiche in cellule viventi contenenti tutti i componenti della via sottostante studiata ha rivelato connessioni nascoste, non osservate prima, nonostante un intenso esame di questa rete. I risultati presentati dimostrano che la strategia PCA ha le caratteristiche necessarie per una convalida generale della funzione genica e una strategia di mappatura della via. Una recente applicazione di un insieme più ampio di PCA ha permesso lo sviluppo di un approccio generale per collegare le azioni dei farmaci su specifiche vie di segnalazione e per rilevare attività impreviste dei farmaci (17).
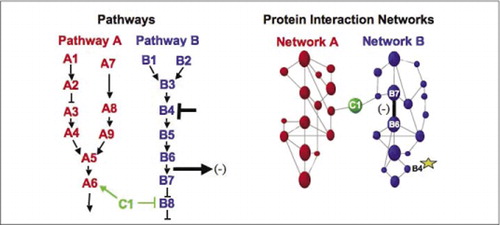
(A sinistra) L’azione di un agente perturbante inibitorio che agisce sulla proteina B4 (T-bar), viene rilevata a valle da un cambiamento nell’interazione delle proteine B6 e B7 l’una con l’altra (freccia). In questo caso, l’effetto della perturbazione è una diminuzione del numero di proteine interagenti (-) come rilevato da un reporter di quell’interazione (segnale di uscita dell’interazione come rilevato da PCA sentinel per esempio). Tuttavia, l’effetto potrebbe essere ugualmente positivo, a seconda delle conseguenze dell’inibizione della proteina a monte. (A destra) All’interno della rete di interazione proteica per la via B, una perturbazione della proteina B4 (stella) si propaga in qualche modo attraverso la rete per influenzare in qualche modo il collegamento (barra larga) tra le proteine B6 e B7. Ciò non implica che la proteina B4 interagisca fisicamente con B6 o B7; la propagazione di un effetto attraverso la rete di interazione proteica può essere dovuta a collegamenti fisici diretti o a processi enzimatici non evidenti nella rete.
Conclusione
Lo sviluppo e l’applicazione di PCA sono ancora in corso. Ad esempio, oltre alle serie limitate, sebbene informative, di applicazioni descritte qui, la strategia viene applicata allo screening su larga scala di interi genomi. Sono stati esplorati problemi più sofisticati di progettazione delle proteine e ripiegamento delle proteine, inclusi studi sui fattori che controllano la selezione delle sequenze per interazioni ottimali tra proteine, proteine e acidi nucleici e proteine e piccole molecole organiche. PCA è un approccio sperimentale molto generale e flessibile, e quindi dovremmo aspettarci di vedere un numero crescente di nuove applicazioni di questo strumento di base alla biologia molecolare e cellulare nel prossimo futuro.
Riconoscimenti
Stephen Michnick detiene il Canada Research Chair in Integrative Genomics. La ricerca citata dal nostro laboratorio è stata finanziata dal Canadian Institutes of Health Research.
- 1. Pelletier, J. N., F. X. Campbell-Valois, e S. W. Michnick. 1998. Oligomerizzazione riassemblaggio diretto al dominio della diidrofolato reduttasi attiva da frammenti progettati razionalmente. Proc. Natl. Acad. Sic. Stati Uniti D’America 95: 12141-12146.Crossref, Medline, CAS, Google Scholar
- 2. Remy, I. e S. W. Michnick. 1999. Selezione clonale e quantificazione in vivo delle interazioni proteiche con test di complementazione proteina-frammento. Proc. Natl. Acad. Sic. Stati Uniti 96: 5394-5399.Crossref, Medline, CAS, Google Scholar
- 3. Il suo nome deriva dal greco antico, che significa “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”. 2002. Test di integrazione dei frammenti proteici beta-lattamasi come sensori in vivo e in vitro delle interazioni proteiche. NAT. Biotecnologia. 20:619–622.Crossref, Medline, CAS, Google Scholar
- 4. Il suo nome deriva dal greco antico, che significa “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”. 2002. Interazioni proteina-proteina monitorate nelle cellule di mammifero attraverso la complementazione di frammenti enzimatici beta-lattamasi. Proc. Natl. Acad. Sic. Stati Uniti 99: 3469-3474.Crossref, Medline, CAS, Google Scholar
- 5. Spotts, J. M., R. E. Dolmetsch, e M. E. Greenberg. 2002. Imaging time-lapse di un’interazione dinamica proteina-proteina dipendente dalla fosforilazione nelle cellule di mammifero. Proc. Natl. Acad. Sic. Stati Uniti D’America 99: 15142-15147.Crossref, Medline, CAS, Google Scholar
- 6. Remy, I. e S. W. Michnick. 2006. Un test di interazione proteina-proteina altamente sensibile basato sulla Gaussia luciferasi. NAT. Metodi 3: 977-979.Crossref, Medline, CAS, Google Scholar
- 7. Ghosh, I., A. D. Hamilton, e L. Regan. 2000. Antiparallel leucine zipper – directed protein reassembly: applicazione alla proteina fluorescente verde. J. Am. Chimica. Soc. 122:5658–5659.Crossref, CAS, Google Scholar
- 8. Hu, C. D., Y. Chinenov, e T. K. Kerppola. 2002. Visualizzazione delle interazioni tra proteine della famiglia bZIP e Rel in cellule viventi mediante complementazione di fluorescenza bimolecolare. Mol. Cella 9: 789-798.Crossref, Medline, CAS, Google Scholar
- 9. Remy, I. e S. W. Michnick. 2004. Una strategia di screening funzionale della libreria cDNA basata su test di complementazione proteica fluorescente per identificare nuovi componenti delle vie di segnalazione. Metodi 32: 381-388.Crossref, Medline, CAS, Google Scholar
- 10. Il suo nome deriva dal greco antico, che significa “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”. 2005. Rilevazione delle interazioni della proteina e screening della biblioteca con i saggi di complementazione del proteina-frammento, p. 637-672. In Interazioni Proteina-Proteina: A Molecular Cloning Manual, 2nd ed. CSH Laboratory Press, Cold Spring Harbor, NY.Google Scholar
- 11. Paulmurugan, R. e S. S. Gambhir. 2003. Monitoraggio delle interazioni proteina-proteina utilizzando la proteina renilla luciferasi sintetica split-frammento-assistita complementazione. Anale. Chimica. 75:1584–1589.Crossref, Medline, CAS, Google Scholar
- 12. Il suo nome deriva dal greco antico, che significa “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra” e “terra”. 2002. Imaging non invasivo delle interazioni proteina-proteina in soggetti viventi utilizzando strategie di complementazione e ricostituzione delle proteine reporter. Proc. Natl. Acad. Sic. Stati Uniti D’America 99: 15608-15613.Crossref, Medline, CAS, Google Scholar
- 13. Il suo nome deriva dal greco antico, che significa “worms”, “Worms”, “Worms”, “Worms”, “Worms”, “Worms”, “Worms”, “Worms”, “Worms”, “Worms”, “Worms”, “Worms”, “Worms” e “Worms”. 2004. Cinetica delle interazioni proteina-proteina regolate rivelate con l’imaging di complementazione della lucciola luciferasi in cellule e animali viventi. Proc. Natl. Acad. Sic. Stati Uniti D’America 101:12288-12293.Crossref, Medline, CAS, Google Scholar
- 14. Il sito utilizza cookie tecnici e di terze parti. 2006. Un mRFP1 migliorato aggiunge il rosso alla complementazione della fluorescenza bimolecolare. NAT. Metodi 3: 597-600.Crossref, Medline, CAS, Google Scholar
- 15. Il suo nome deriva da quello di una donna di nome S. W. Michnick. 2004. PKB / Akt modula la segnalazione TGF-beta attraverso un’interazione diretta con Smad3. NAT. Biol cellulare. 6:358–365.Crossref, Medline, CAS, Google Scholar
- 16. Remy, I. e S. W. Michnick. 2001. Visualizzazione di reti biochimiche nelle cellule viventi. Proc. Natl. Acad. Sic. Stati Uniti D’America 98: 7678-7683.Crossref, Medline, CAS, Google Scholar
- 17. Il suo nome deriva dal greco antico, che significa “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”.. 2006. Identificare effetti fuori bersaglio e fenotipi nascosti di farmaci nelle cellule umane. NAT. Chimica. Biol. 2:329–337.Crossref, Medline, CAS, Google Scholar
- 18. Pelletier, J. N., K. M. Arndt, A. Pluckthun, e S. W. Michnick. 1999. Una selezione in vivo library-versus-library di interazioni proteina-proteina ottimizzate. NAT. Biotecnologia. 17:683–690.Crossref, Medline, CAS, Google Scholar
- 19. Remy, I., I. A. Wilson, e S. W. Michnick. 1999. Attivazione del recettore dell ‘ eritropoietina mediante un cambiamento di conformazione indotto dal ligando. Scienza 283: 990-993.Crossref, Medline, CAS, Google Scholar
- 20. Gegg, C. V., K. E. Bowers, e C. R. Matthews. 1997. Sondare unità di piegatura indipendenti minime in diidrofolato reduttasi per dissezione molecolare. Proteina Sic. 6:1885–1892.Crossref, Medline, CAS, Google Scholar
- 21. Il suo nome deriva dal greco antico. 1997. Monitoraggio delle interazioni proteina-proteina nelle cellule eucariotiche intatte mediante integrazione di beta-galattosidasi. Proc. Natl. Acad. Sic. Stati Uniti D’America 94: 8405-8410.Crossref, Medline, CAS, Google Scholar
- 22. Il suo nome deriva dal greco Oz 2000. Un indicatore fluorescente per la rilevazione delle interazioni proteina-proteina in vivo basato sull’impionbatura della proteina. Anale. Chimica. 72:5151–5157.Crossref, Medline, CAS, Google Scholar
- 23. Il sito utilizza cookie tecnici e cookie di profilazione di terze parti. 2005. Rilevamento delle interazioni proteina-proteina con una trappola di riassemblaggio del frammento proteico fluorescente verde: scopo e meccanismo. J. Am. Chimica. Soc. 127:146–157.Crossref, Medline, CAS, Google Scholar
- 24. Nyfeler, B., S. W. Michnick, e H. P. Hauri. 2005. Catturare le interazioni proteiche nella via secretoria delle cellule viventi. Proc. Natl. Acad. Sic. Stati Uniti D’America 102: 6350-6355.Crossref, Medline, CAS, Google Scholar
- 25. Arndt, K. M., J. N. Pelletier, K. M. Muller, T. Alber P.S. Michnick, e A. Pluckthun. 2000. Una coppia di peptidi a spirale eterodimerica selezionata in vivo da un insieme libreria-versus-libreria progettato. J. Mol. Biol. 295:627–639.Crossref, Medline, CAS, Google Scholar
- 26. Remy, I. e S. W. Michnick. 2004. Regolazione dell’apoptosi da parte della proteina Ft1, un nuovo modulatore della protein chinasi B / Akt. Mol. Cellula. Biol. 24:1493–1504.Crossref, Medline, CAS, Google Scholar
- 27. Il sito utilizza cookie tecnici e di terze parti per migliorare la tua esperienza di navigazione. 2006. Uno schermo basato a retrovirus-della prova di complementazione della proteina rivela i partner funzionali AKT1-leganti. Proc. Natl. Acad. Sic. USA 103:15014–15019.Crossref, Medline, CAS, Google Scholar
- 28. Cody, V., J.R. Luft, E. Ciszak, T.I. Kalman, and J.H. Freisheim. 1992. Crystal structure determination at 2.3 A of recombinant human dihydrofolate reductase ternary complex with NADPH and methotrexate-gamma-tetrazole. Anticancer Drug Des. 7:483–491.Medline, CAS, Google Scholar