Articolo
Josivan Gomes Lima1*, Marcel Catão Ferreira dos Santos1, Julliane Tamara Araújo de Melo Campos2
1Dipartamento de medicina clínica, disciplina de endocrinologia e metabologia. Hospital Universitário Onofre Lopes, Universidade Federal do Rio Grande do Norte (UFRN), Natal, RN, Brasile
2Faculty of Health Sciences of Trairi, Federal University of Rio Grande do North (UFRN), Natal, RN, Brasile
Abstract
La lipodistrofia generalizzata congenita (CGL) è una rara e grave malattia autosomica malattia recessiva. I pazienti sono difettosi nello stoccaggio del grasso corporeo e, di conseguenza, depositano il grasso nei tessuti ectopici, principalmente nel fegato, e possono sviluppare cirrosi. La resistenza all’insulina è un risultato tipico, causando il diabete che richiede alte dosi giornaliere di insulina. Nello stato di Rio Grande do Norte, Brasile, abbiamo una delle più grandi coorti di pazienti con CGL. In questo articolo, esaminiamo la fisiopatologia, il quadro clinico e il trattamento di questa malattia.
Introduzione
Il diabete di tipo 2 è un problema di salute mondiale e di solito deriva dal peso eccessivo e dall’aumento del grasso viscerale che causa la resistenza all’insulina periferica e l’incapacità del pancreas di rilasciare insulina per compensare questa resistenza. Altri tipi meno comuni di diabete si verificano a causa di specifiche mutazioni genetiche, come la Lipodistrofia generalizzata congenita (CGL), nota anche come Lipodistrofia congenita di Berardinelli-Seip (BSCL). La CGL è una malattia autosomica recessiva che è classificata in quattro tipi, basati sulla mutazione del gene. I geni alterati svolgono funzioni essenziali per la formazione di adipociti, la produzione di lipidi e la corretta conservazione all’interno dell’adipocita. Le mutazioni diminuiscono il tessuto adiposo con conseguente deposizione di grasso in siti ectopici, causando fegato grasso, alterato metabolismo dei carboidrati, grave insulino-resistenza con iperinsulinemia e caratteristiche acromegaloidi e dislipidemia1-3. La sindrome CGL ha circa 500 casi segnalati nel mondo. In Brasile, nello Stato di Rio Grande do Norte (RN), abbiamo diagnosticato, trattato e seguito 54 casi negli ultimi 20 anni4, 5. In uno studio descrittivo utilizzando dati secondari, abbiamo stimato un totale di 103 pazienti in RN6. Ciò indica una prevalenza molto più elevata di quella riportata in letteratura (1: 1 milione) 7.
Formazione e stoccaggio di triacilglicerolo in goccioline lipidiche
La biosintesi dei trigliceridi e dei fosfolipidi (Figura 1A) inizia con glicerolo-3-fosfato aciltransferasi (GPAT) che acilizza il glicerolo-3-fosfato in posizione 1, formando 1-acilglicerolo-3-fosfato (acido lisofosfatidico). È seguita da un’altra fase di acilazione in posizione due da parte dell’enzima AGPAT (1-acilglicerolo-3-fosfato aciltransferasi), originario di 1,2-Diacilglicerolo-3-fosfato (acido fosfatidico). È un punto intermedio chiave nella via della biosintesi sia dei trigliceridi che dei fosfogliceridi. Esistono 11 isoforme di enzimi AGPAT, codificate da diversi geni4. AGPAT1 e AGPAT2 sono i più ampiamente studiati. AGPAT1 è presente ad alti livelli nel testicolo, nel pancreas e, in misura minore, nel tessuto adiposo e in altri tessuti come cuore, placenta, cervello, polmone, mentre AGPAT2 è abbondante nel tessuto adiposo. Nei seguenti passaggi, l’enzima citosolico fosfatidico fosfatasi acida (PAP o lipina) origina 1,2-diacilglicerolo e la 1,2-diacilglicerolo aciltransferasi (DGAT) forma triacilglicerol4. L’acido fosfatidico e il diacilglicerolo possono anche originare altri fosfolipidi come cardiolipina, fosfatidilinositolo e fosfatidilcolina.
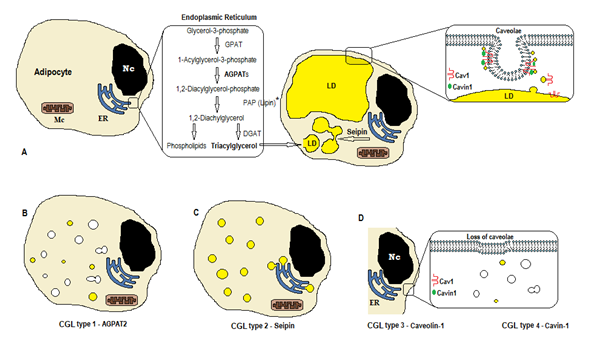
Figura 1. Schema di sintesi dei trigliceridi secondo i tipi CGL. (A) Normale sintesi e conservazione del triacilglicerolo (TAG) nell’adipocita. (B) La mutazione di AGPAT2 diminuisce la produzione di TAG (alcuni sono ancora sintetizzati sotto stimolazione di altri AGPATs). (C) Mutazione del gene seipin diminuzione TAG sintesi e lipid droplet (LD) formazione e fusione. (D) Caveolin-1 e Cavin-1 sono necessari per la formazione e la stabilizzazione delle caveole. La mutazione in CAV1 (tipo 3) o CAVIN1 (tipo 4) può causare la perdita di caveole nella membrana. Nc, nucleo. Reticolo endoplasmatico. Mc, mitocondri. * La lipina è un enzima citosolico ancorato dalla seipina nel pronto soccorso.
Tali reazioni si verificano nel reticolo endoplasmatico degli adipociti (ER), dove un progressivo accumulo di trigliceridi provoca la formazione di piccole goccioline lipidiche (LD)8. Il prodotto del gene BSCL2 è una proteina transmembrana chiamata seipin che causa la fusione di piccole LD, originando grandi LD. Seipin risiede nel ER e si concentra alla giunzione con LD nascente, facilitando il traffico lipidico tra ER e LD e l’incorporazione di trigliceridi in LD9. Seipin può anche fungere da ancoraggio ER all’enzima citosolico lipina 1. Oltre ad essere necessario per la fusione delle gocce lipidiche, le dimensioni e la morfologia, seipin è anche essenziale per l’adipogenesi (tramite interazione con lipina 1) e la lipolisi dei trigliceridi cellulari10, 11. La carenza di seipin ostacola la differenziazione dei pre-adipociti in adipociti e influenza la maturazione finale9, come dimostrato da studi su cellule staminali mesenchimali con BSCL2 eliminato 12. I tessuti non adiposi esprimono anche seipin e altre funzioni devono essere determinate.
Negli adipociti, le caveole, che sono invaginazioni specializzate della membrana 50-100nm, rappresentano il 20% dell’area della membrana plasmatica, rendendo gli adipociti le cellule con la più alta densità di caveole13. La formazione di goccioline lipidiche richiede una proteina di membrana (Caveolina – il componente principale delle membrane caveolae) e una proteina citoplasmatica (Cavina-1)14. I geni CAV1, CAV2 e CAV3 codificano tre forme di caveolina con strutture simili (Caveolina-1, Caveolina-2 e Caveolina-3, rispettivamente). Caveolin-1 e Caveolin-2 sono presenti negli adipociti, fibroblasti e cellule endoteliali e Caveolin-3 è presente solo nel muscolo scheletrico e cardiaco13, 15. Caveolin-1 è il più importante e il più studiato. È espresso in due diverse isoforme (1a e 1b). Caveolin-1 trasloca dalla membrana plasmatica alla goccia lipidica, essendo necessario per il traffico lipidico e il metabolismo16. Le goccioline lipidiche immagazzinano i trigliceridi dopo l’alimentazione e queste molecole vengono idrolizzate in acido grasso e rilasciate durante il digiuno; questo meccanismo può essere regolato da Caveolin-116. La carenza di Caveolin-1 aumenta anche la suscettibilità alla morte cellulare da parte dell’autofagia17.
Il gene CAVIN1 codifica una proteina citoplasmatica chiamata proteina associata alle caveole 1 (Cavin-1)14, 16, che è obbligatoria per la formazione e la stabilizzazione delle caveole. La cavina-1 è espressa in adipociti, cellule muscolari e altre cellule ed è anche essenziale nella trasmissione di segnali originati dalle caveole14, 18. Il knockout del gene CAV1 causa una mancanza di caveole nelle cellule non muscolari, mentre il knockout di CAVIN1 causa l’assenza di caveole in tutti i tessuti, incluso il muscolo14. La mancanza di caveole può influenzare la regolazione della lipolisi, del flusso di acidi grassi, della sintesi dei trigliceridi e dei segnali di altre vie.
Tipi di CGL
Sulla base di alterazioni genetiche rilevabili, sono descritti quattro tipi. I tipi 1 e 2 sono responsabili di oltre il 95% dei casi e il tipo 2 ha un fenotipo più gravemente colpito. Sono stati segnalati solo un caso di tipo 3 e circa 30 casi di tipo 44.
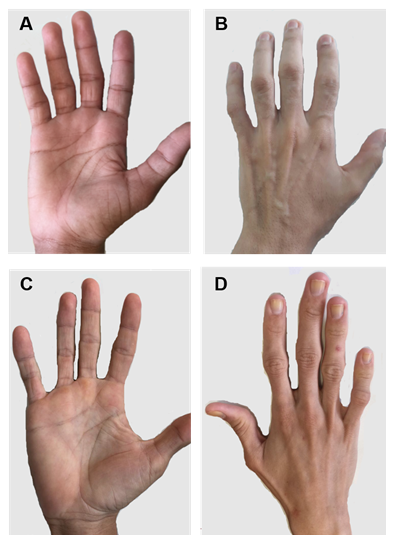
Figura 2. Mani di pazienti con CGL di tipo 1 e 2. (A) e (B) Vista anteriore e posteriore delle mani di pazienti di tipo 1. Mani apparentemente normali, poiché c’è ancora tessuto grasso meccanico. (C) e (D) Vista anteriore e posteriore delle mani dei pazienti di tipo 2. La gravità della malattia è maggiore e la mancanza di grasso è evidente e facilmente evidente.
CGL Tipo 1. Nel 1999, Garg et al. descritto mutazione dei pazienti sul cromosoma 9q34, e tre anni dopo Agarwal et al. ha mostrato AGPAT2 come l’enzima affetto da questa mutazione2, 19. A causa della mutazione di questo AGPAT2, nessuna o minima produzione di triacilglicerolo avviene dallo stimolo di altre isoforme. Il fenotipo dei topi knockout AGPAT2 è simile a quello degli esseri umani con tipo CGL, confermando il ruolo di questo enzima nella fisiopatiologia20, 21.
CGL Tipo 2. Magre et al. sono stati i primi a identificare la mutazione nel gene seipin (cromosoma 11q13) 3. Le mutazioni (per lo più sciocchezze) del gene seipin (BSCL2) producono una proteina troncata e possono influenzare il metabolismo lipidico con diversi meccanismi: a) diminuzione della stabilità della seipina; b) riduzione della capacità di legare la lipina 1; e c) incapacità di oligomerizzare e localizzarsi esclusivamente alla membrana er11. Alcune cellule sono ancora in grado di generare triacilglicerolo e piccole goccioline lipidiche, ma grandi goccioline lipidiche sono assenti a causa della perdita della capacità di fusione di queste piccole goccioline lipidiche. C’è anche un fallimento nell’espressione di fattori adipogeni, come il recettore gamma attivato dal proliferatore del perossisoma (PPARG), così come l’adiponectina e la proteina legante l’acido grasso degli adipociti (FABP4)11, 16. La carenza di Seipin altera l’adipogenesi, aumenta la lipolisi e previene l’accumulo di trigliceridi negli adipociti.
CGL tipo 3. Questo tipo è stato descritto recentemente in un paziente che pur avendo fenotipo CGL non ha avuto mutazioni nei geni AGPAT2 o BSCL222. I topi con una mutazione in Cav1 sono resistenti all’obesità indotta dalla dieta e hanno insulino-resistenza, ipertrigliceridemia, diminuzione dell’adiponectina, riduzione della massa grassa e piccoli adipociti16. Dopo aver scelto i geni candidati sulla base di studi sui topi, Kim et al. confermata la presenza di una mutazione senza senso nel gene caveolin-1 (CAV1), sul cromosoma 7q3122.
CGL tipo 4. In questo è un tipo raro il gene interessato è il CAVIN1, che codifica la proteina Cavin-1. Negli esseri umani, è stato riportato in pazienti con lipodistrofia congenita generalizzata e distrofia muscolare15, 23.
Recentemente sono state descritte anche mutazioni nei geni PCYT1A e PPARG che causano la lipodistrofia24, 25.
Caratteristiche cliniche
I pazienti con CGL presentano solitamente facies acromegaloidi, acantosi nigricans, febomegalia, epatomegalia e ipertrofia muscolare5, 26, 27. Diversi autori citano l’ernia ombelicale come scoperta clinica della sindrome26. Ne abbiamo valutato la frequenza nella nostra serie di pazienti e nessuno di loro ha presentato questo cambiamento28. Infatti, l’assenza di tessuto adiposo periumbilico causa la protrusione della cicatrice ombelicale, e questo può essere erroneamente diagnosticato come un’ernia28, 29.
Una volta che gli adipociti non possono immagazzinare adeguatamente il grasso, si accumula in altri tessuti come fegato e muscoli, causando una grave resistenza all’insulina. La densitometria ossea (DXA) può mostrare una densità minerale ossea normale o alta30 e una riduzione del grasso corporeo totale (di solito inferiore al 6%)27. Come conseguenza del basso grasso corporeo, anche l’adiponectina e la leptina sieriche sono basse27. Poiché la leptina è essenziale nel controllo della fame, questi pazienti hanno tipicamente iperfagia, che è facilmente evidente fin dall’infanzia. L’adiponectina svolge un ruolo importante come sensibilizzatore all’insulina e la sua mancanza peggiora la resistenza all’insulina. Nonostante questo, inizialmente, il glucosio e l’emoglobina glicata sono normali a scapito di livelli di insulina molto alti. Il diabete di solito inizia alla pubertà; nella nostra serie, l’età media di insorgenza era di 15,8±7,1 anni27. Inizialmente, sono controllati con farmaci orali, che necessitano di alte dosi di insulina in pochi anni27. L’ipertensione arteriosa si verifica in un terzo dei pazienti27.
Esistono alcune caratteristiche cliniche specifiche di ciascun tipo di CGL. I pazienti con tipo 1 presentano ancora grasso adiposo meccanico, specialmente nelle palme, nelle suole, nelle regioni orbitali e peri-articolari31. Al contrario, i pazienti di tipo 2 mostrano un’assenza di tessuti grassi metabolici e meccanici. Seipin è altamente espresso nel cervello e nel cervelletto ed è anche coinvolto nella regolazione delle funzioni neurali. Più della metà dei pazienti di tipo 2 ha un deficit cognitivo1, 8. I tipi 3 e 4 hanno la conservazione del grasso meccanico e del midollo osseo e il tipo 4 ha debolezza muscolare associata ad alta creatina chinasi sierica e instabilità spinali 15.
Esistono anche caratteristiche cliniche specifiche per genere. Le ovaie policistiche e l’amenorrea sono comuni32. I cicli mestruali di solito tornano alla normalità con l’uso di metreleptina, probabilmente a causa del miglioramento della sensibilità all’insulina e del ripristino della pulsatilità LH32. Gli uomini di tipo 2 possono avere teratozoospermia a causa della mancanza di seipin nelle cellule germinali33.
L’ipertrigliceridemia si verifica fin dai primi anni di vita e può causare pancreatite acuta. L’HDL è solitamente inferiore a 30 mg / dL. Elevazioni degli enzimi epatici è anche una scoperta precoce e provengono dalla deposizione di grasso nel fegato. La progressiva riduzione delle piastrine sieriche suggerisce un peggioramento della malattia epatica e una probabile cirrosi34.
Poiché la Cavina-1 è presente nelle cellule muscolari, i pazienti con tipo 4 hanno una lieve debolezza muscolare e una creatina chinase15 elevata.
L’aspettativa di vita, principalmente nel tipo 2, è sostanzialmente diminuita, con la morte non di rado si verifica prima dei 30 anni di età (esperienza personale basata su 20 pazienti deceduti negli ultimi 19 anni). Le cause di morte sono correlate al diabete (insufficienza renale, morte improvvisa), al fegato (cirrosi, sanguinamento digestivo) o alle infezioni.
Diagnosi e trattamento
La diagnosi CGL si basa su dati clinici: caratteristiche acromegaloidi, acantosi nigricana, riduzione del grasso corporeo totale, ipertrofia muscolare e protrusione della cicatrice ombelicale. Inoltre, i dati di laboratorio possono mostrare il diabete con grave insulino-resistenza e ipertrigliceridemia. I test di imaging possono aiutare a identificare i depositi ectopici di grasso principalmente nel fegato e nel pancreas (steatosi epatica con epatomegalia e steatosi pancreatica). Il DXA può confermare il basso grasso corporeo e l’alta densità ossea30.
Il trattamento della CGL consiste in uno stretto controllo della dieta con la diminuzione dell’assunzione di grassi, principalmente trigliceridi e alimenti ad alto indice glicemico per prevenire e controllare le comorbidità29. Tuttavia, la dieta ideale è un obiettivo impegnativo da raggiungere a causa dell’aumento dell’appetito e della severa restrizione sostenuta. L’attività fisica deve essere incoraggiata anche per migliorare il controllo delle comorbidità, tranne nei pazienti con controindicazioni come la cardiomiopatia grave29.
Per quanto riguarda il trattamento farmacologico, questi pazienti possono essere trattati con i soliti farmaci per il diabete, l’ipertensione e le linee guida sulla dislipidemia. La prima scelta per il trattamento del diabete e della resistenza all’insulina è la metformina, ma di solito non è sufficiente. A differenza del trattamento della lipodistrofia parziale, i tiazolidinedioni devono essere usati con caution29. Vengono utilizzati altri agenti antidiabetici orali, ma non sono stati specificamente studiati nei pazienti con CGL. Esistono dati sugli animali che suggeriscono che l’uso di inibitori SGLT2 (dapagliflozin) potrebbe avere benefici nella prevenzione della cardiomiopatia35; sono necessari studi per confermarlo nell’uomo. Man mano che la malattia progredisce e si verifica una grave insulino-resistenza, sono necessarie alte dosi giornaliere di insulina. La mancanza di tessuto adiposo sottocutaneo è un problema nella somministrazione di alte dosi di insulina. Può essere necessaria una maggiore concentrazione di insulina (U-300 o U500) 36. Questi pazienti presentano grave dislipidemia, principalmente a causa dell’aumento dei trigliceridi e della bassa HDL, e quindi l’uso del fibrato è talvolta necessario per prevenire la pancreatite acuta. Inoltre, a causa dell’elevato rischio cardiovascolare di questi pazienti, deve essere preso in considerazione l’intervento con una statina e gli obiettivi di LDL o non HDL devono essere rigorosi29.
Le iniezioni giornaliere di metreleptina causano una significativa diminuzione dell’appetito e apportano benefici abbassando la glicemia, la trigliceridemia e gli enzimi epatici. È notevole, soprattutto nei bambini, la riduzione della circonferenza addominale, probabilmente dovuta ad una riduzione dell’epatomegalia.
Conclusione
La CGL è una malattia rara e grave che può verificarsi con il diabete (che di solito richiede alte dosi di insulina) e la morte precoce. Il fenotipo del paziente è abbastanza caratteristico, richiedendo, tuttavia, la conoscenza della sindrome da parte degli operatori sanitari per fare una diagnosi precoce. Metreleptin sembra essere l’unico farmaco al momento in grado di modificare la storia naturale della malattia.
Conflitto di interessi: nessuno.
- Nolis T. Esplorare la fisiopatologia dietro le più comuni lipodistrofie genetiche e acquisite. Rivista di genetica umana. 2014 Gennaio; 59 (1): 16-23.
- Agarwal AK, Arioglu E, De Almeida S, et al. AGPAT2 è mutato nella lipodistrofia generalizzata congenita legata al cromosoma 9q34. Nat Genet. 2002 Maggio; 31(1): 21-3.
- Magre J, Delepine M, Khallouf E, et al. Identificazione del gene alterato nella lipodistrofia congenita di Berardinelli-Seip sul cromosoma 11q13. Natura genetica. 2001 Agosto; 28 (4): 365-70.
- Patni N, Garg A. Lipodistrofie generalizzate congenite new nuove conoscenze sulle disfunzioni metaboliche. Recensioni di natura Endocrinologia. 2015 Settembre; 11 (9): 522-34.
- Garg A. Lipodistrofie acquisite ed ereditate. Il New England journal of medicine. 2004 Mar 18; 350 (12): 1220-34.
- de Azevedo Medeiros LB, Candido Dantas VK, Craveiro Sarmento AS, et al. Alta prevalenza di lipodistrofia congenita di Berardinelli-Seip nello Stato di Rio Grande do Norte, Brasile nord-orientale. Diabetol Metab Syndr. 2017; 9: 80.
- Chiquette E, Oral EA, Garg A, et al. Stima della prevalenza della lipodistrofia generalizzata e parziale: risultati e sfide. Diabete, Sindrome metabolica e obesità: target e terapia. 2017: 375-83.
- Wee K, Yang W, Sugii S, et al. Verso una comprensione meccanicistica della lipodistrofia e delle funzioni di seipin. Rapporti di bioscienza. 2014; 34(5).
- Dollet L, Magre J, Cariou B, et al. Funzione di seipin: nuove intuizioni dai modelli di mouse knockout Bscl2 / seipin. Biochimie. 2014 Gennaio; 96: 166-72.
- Sim MF, Dennis RJ, Aubry EM, et al. La proteina umana seipin della lipodistrofia è un adattatore della membrana di ER per la lipina adipogena della fosfatasi di PA 1. Metabolismo molecolare. 2012; 2(1): 38-46.
- Sim MF, Talukder MM, Dennis RJ, et al. L’analisi delle mutazioni naturali nella proteina lipodistrofia umana seipin rivela molteplici potenziali meccanismi patogeni. Diabetologia. 2013 Novembre; 56 (11): 2498-506.
- Payne VA, Grimsey N, Tuthill A, et al. Il gene umano della lipodistrofia BSCL2 / seipin può essere essenziale per la differenziazione normale degli adipociti. Diabete. 2008 Agosto; 57 (8): 2055-60.
- Cohen AW, Hnasko R, Schubert W, et al. Ruolo delle caveole e delle caveoline nella salute e nella malattia. Recensioni fisiologiche. 2004 Ottobre; 84 (4): 1341-79.
- Pilch PF, Liu L. Grotte grasse: caveole, traffico lipidico e metabolismo lipidico negli adipociti. Tendenze in endocrinologia e metabolismo: TEM. 2011 Agosto; 22 (8): 318-24.
- Hayashi YK, Matsuda C, Ogawa M, et al. Le mutazioni PTRF umane causano una carenza secondaria di caveoline con conseguente distrofia muscolare con lipodistrofia generalizzata. J Clin Investire. 2009 Settembre; 119 (9): 2623-33.
- Parton RG, del Pozo MA. Caveolae come sensori a membrana plasmatica, protettori e organizzatori. Nature recensisce Biologia cellulare molecolare. 2013 Febbraio; 14 (2): 98-112.
- Le Lay S, Briand N, Blouin CM, et al. Il modello lipoatrofico caveolin-1 carente del topo rivela l’autofagia in adipocytes maturi. Autofagia. 2010 Agosto; 6 (6): 754-63.
- Liu L, Brown D, McKee M, et al. La cancellazione di Cavin / PTRF causa la perdita globale di caveole, dislipidemia e intolleranza al glucosio. Metabolismo cellulare. 2008 Ottobre; 8 (4): 310-7.
- Garg A, Wilson R, Barnes R, et al. Un gene per la lipodistrofia generalizzata congenita mappa al cromosoma umano 9q34. Il Journal of clinical endocrinology and metabolism. 1999 Settembre; 84 (9): 3390-4.
- Vogel P, Leggi R, Hansen G, et al. Patologia della lipodistrofia generalizzata congenita nei topi Agpat2 -/ -. Patologia veterinaria. 2011 Maggio; 48 (3): 642-54.
- Cortes VA, Curtis DE, Sukumaran S, et al. Meccanismi molecolari di steatosi epatica e insulino-resistenza nel modello murino AGPAT2-carente di lipodistrofia generalizzata congenita. Metabolismo cellulare. 2009 Febbraio; 9 (2): 165-76.
- Kim CA, Delepine M, Boutet E, et al. Associazione di una mutazione omozigote nonsense caveolin-1 con lipodistrofia congenita di Berardinelli-Seip. J Clin Endocrinol Metab. 2008 Aprile; 93 (4): 1129-34.
- Rajab A, Straub V, McCann LJ, et al. Aritmia cardiaca fatale e sindrome del QT lungo in una nuova forma di lipodistrofia generalizzata congenita con increspatura muscolare (CGL4) dovuta a mutazioni di PTRF-CAVINA. PLoS genetics. 2010 Mar 12; 6 (3): e1000874.
- Payne F, Lim K, Girousse A, et al. Mutazioni che interrompono la via della fosfatidilcolina di Kennedy negli esseri umani con lipodistrofia congenita e malattia del fegato grasso. Proc Natl Acad Sci U S A. 2014 Giugno 17; 111 (24): 8901-6.
- Dyment DA, Gibson WT, Huang L, et al. Le mutazioni bialleliche a PPARG causano una lipodistrofia congenita generalizzata simile alla sindrome di Berardinelli-Seip. Eur J Med Genet. 2014 Settembre; 57 (9): 524-6.
- Garg A. Revisione clinica#: Lipodistrofie: disturbi genetici e acquisiti del grasso corporeo. Il Journal of clinical endocrinology and metabolism. 2011 Novembre; 96 (11): 3313-25.
- Lima JG, Nobrega LH, de Lima NN, et al. Dati clinici e di laboratorio di una vasta serie di pazienti con lipodistrofia generalizzata congenita. Diabetol Metab Syndr. 2016; 8: 23.
- Lima GJ, Lima NN, Oliveira CF, et al. Ernia ombelicale nei pazienti con sindrome di Berardinelliseip: è davvero ernia. J Clin Mol Endocrinol. 2015; 1(1): 3.
- Brown RJ, Araujo-Vilar D, Cheung PT, et al. La diagnosi e la gestione delle sindromi lipodistrofiche: una linea guida pratica multi-società. J Clin Endocrinol Metab. 2016 Dicembre; 101 (12): 4500-11.
- Lima JG, Nobrega LH, Lima NN, et al. La densità ossea nei pazienti con lipodistrofia congenita di Berardinelli-Seip è maggiore nei siti trabecolari e nei pazienti di tipo 2. J Clin Densitom. 2016 Novembre 25.
- Simha V, Garg A. Eterogeneità fenotipica nella distribuzione del grasso corporeo in pazienti con lipodistrofia generalizzata congenita causata da mutazioni nei geni AGPAT2 o seipin. J Clin Endocrinol Metab. 2003 Novembre; 88 (11): 5433-7.
- Musso C, Cochran E, Javor E, et al. L’effetto a lungo termine della terapia con leptina umana metionil ricombinante sull’iperandrogenismo e sulla funzione mestruale nella funzione femminile e ipofisaria nei pazienti lipodistrofici ipoleptinemici maschili e femminili. Metabolismo. 2005 Febbraio; 54 (2): 255-63.
- Jiang M, Gao M, Wu C, et al. La mancanza di seipin testicolare causa la sindrome di teratozoospermia negli uomini. Proc Natl Acad Sci U S A. 2014 Maggio 13; 111(19): 7054-9.
- Mitchell O, Feldman DM, Diakow M, et al. La fisiopatologia della trombocitopenia nella malattia epatica cronica. Epatomed. 2016; 8: 39-50.
- Joubert M, Jagu B, Montaigne D, et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes. 2017 Apr; 66(4): 1030-40.
- Lima JG, Lima NN, Lima RLM, et al. Glargine U300 Insulin as a Better Option than Degludec U100 to Treat a Congenital Generalized Lipodystrophy Patient. Clin Diabetes Res. 2017; 1(1).