小児科におけるFrontiers
- はじめに
- QT延長症候群
- KCNQ1遺伝子の変異は、すべてのLQTSの中で最も一般的な形態であり、LQTS1型(LQTS1)と呼ばれる。 すべてのLQTS原因突然変異の50%がKCNQ1遺伝子に見出される(20)。 Kvlqt1(Kv7.1とも呼ばれる)は、KCNQ1遺伝子によって作られたタンパク質であり、細胞内の小胞体に四量体を形成し、kcne1によってコードされるタンパク質ミンクと推定上共集合し、心筋細胞の原形質膜に輸送され、そこで心臓組織の活動電位の再分極を促進するゆっくりと活性化する電流を媒介し、この電流はIKsとして知られています(図1)。 LQTS1原因KCNQ1変異は、主にミスセンス変異であり、まれに、彼らはC末端領域(におけるフレームシフト変異である可能性があります23)。 KCNQ1変異キャリアにおける心臓不整脈は、アドレナリン作動性ドライブ、例えば、感情的ストレス、身体運動、ダイビング、水泳(24-26)によってトリガされます。 Kvlqt1の膜貫通ドメインに変異を有する患者は、LQTS関連の心臓イベントのリスクが高く、交感神経刺激に対する感受性が高い(26、27)。 ミスセンス変異を有する患者は、非センスまたは切り捨て変異を有する患者と比較してリスクが高い(27、28)。 KCNQ1遺伝子における3′-UTRの変化はまた、おそらく遺伝子の発現に影響を与えることによって、不整脈感受性に有意に影響を与える(29)。 KCNQ1突然変異による不整脈の患者はβ遮断薬にかなりよく答えますが、何人かの患者はまだこの薬物に対してより少なく敏感また更に抵抗力があ 最近の記事では、Barsheshet et al. (30)は、Kvlqt1の細胞質ループ(cループ)領域の外側に変異を有する患者は、β遮断薬に応答性が低いと主張した。 1 図1. 心室活動電位に寄与するイオン電流(A)および心筋細胞の模式図(のみ)遺伝性不整脈症候群(B)の病因に関与するタンパク質を表示する。 (A)では、活動電位はECGの間の行為のおおよその時間と一直線に並ぶ。 (B)では、qt延長症候群4型に関与するアダプタータンパク質であるankyrin-Bは描かれていない。
- LQTS2
- LQTS3
- LQTS4
- LQTS5
- LQTS6
- LQTS7
- Lqts8
- LQTS9およびLQTS10
- LQTS11
- lqts12
- LQTS13
- 後天性LQTS
- QT延長症候群の三つの一般的な形態における心電図的特徴
- 遺伝子型-表現型
- LQTSの臨床管理
- : サウジの視点
- 利益相反に関する声明
- 謝辞
はじめに
家族性または遺伝性心臓不整脈は、不整脈の有意な割合を占め、また突然の心臓死(SCD)にも原因となる(1、2)。 最後の二十年の間に、科学者および臨床医は生来の家族性不整脈(1-8)の複雑で、複雑なメカニズムを解明するための巨大な努力を提供しました。 不整脈発生のメカニズムを理解するためには、心臓の細胞構造とその電気生理学的性質の基礎を知る必要があります。 心筋細胞は心臓の主要な機能細胞であり、インパルスが迅速かつ均一に伝播するようにそれらは広範囲に結合されている。 心筋細胞はインターカレートディスクと呼ばれる特殊な境界によって互いに分離され、ギャップ接合タンパク質、心臓デスモソーム、イオンチャネルはインターカレートディスクに位置する(9)。 ギャップ接合は、小分子の細胞間交換を可能にし、また、一つの細胞からその隣接する細胞に興奮電流を流すことを可能にする密に充填されたコネク Desmosomesはadherensの接続点と共に個々の心筋細胞の機械付属品に責任があります。 挿入されたディスク内のこれらの成分はすべて順番に分離され、各成分はその独特の機能を発揮する;1つの成分の破壊は他の成分の機能に影響し、心臓が不整脈を発症する素因となる(9-11)。 心臓イオンチャネルは、心臓リズムの生成と伝播のために不可欠な、細胞膜全体のイオン電流の電圧ゲートと複雑に調整された内側と外側の動きを提 QT延長症候群(LQTS),qt短縮症候群(SQTS),副鼻腔症候群(sss),心臓伝導欠損症(CCD),Brs,カテコールアミン作動性多型心室頻拍(CPVT),早期再分極症候群(Γ),家族性心房細動(A F)は現在知られている心臓チャネロパチーであり,心臓リズムの生成と伝播に関連する遺伝子の単一または複数の欠損のために起こる可能性がある。
このレビューでは、LQTSリンク不整脈、その病態生理および現在利用可能な臨床管理についてのみ説明します。 我々は、サウジアラビア(1、12-14)の家族性不整脈における遺伝病理の解明に先駆的になっている、我々はまだサウジアラビアで心原性調査を行っている、このレビューの終わりに、我々は失神とSCDの歴史を持ついくつかのサウジアラビアの家族から得られた遺伝的および臨床所見について現在利用可能な知識を議論します。 また、リヤドのKing Faisal Specialist Hospital and Research Centreのチームからlqtsに関する最近公開されたデータを追加します(15)。
QT延長症候群
先天性LQTSは、心電図(ECG)上のQT間隔の延長によって定義される遺伝性疾患である。 すべての形態のLQTを有する患者は、心室頻脈性不整脈、再発性失神を引き起こすtorsades de pointes(TdP)、またはSCDの素因がある。 多くの場合、失神または突然死が最初で唯一の症状である可能性があります。 LQTSは、世界中の推定1で2,000人に影響を与えます(16)。 LQTSの特徴は、ECG上のQT間隔の延長(心拍数、すなわちQtcに対して補正される)である。 QTcの正常値は、男性では440ms、女性では450msである。 子供では、年齢と性別に依存する値が関連しています。 LQTS診断のための最近のコンセンサス勧告は次のとおりです(17, 18):
1. LQTSが診断される:
a.LQTSリスクスコア≥3.5の存在下でQT延長の二次的原因がない場合および/または
b.LQTS遺伝子のいずれかまたは
c. Bazettの式(QTc)を使用して心拍数を補正したQT間隔の存在下で、反復した500リード心電図において、QT延長の二次的原因がない場合には、qt延長の二次的原因が
2. LQTSは、QT延長の二次的原因がない場合および病原性変異がない場合に、原因不明の失神を有する患者において、480および499msの間のQTcの存在下で12リード
qtc期間が500ms以上の患者は、QTc期間<500msの患者と比較して、出生から20歳までの患者(19)と比較して、最初の失神を経験する累積確率が有意に高かった。 しかし、2、3、および4失神エピソードを経験した患者は、その後の失神エピソードのリスクは、狭いまたは長期のQTc期間(持っていた患者の間で事実上同一 LQTSの分子基盤は不均一であり、現在までに、13の異なる遺伝子の変異がLQTSの原因であると記載されている(1-8、19、20)。 遺伝的欠陥は、通常、これらの13の遺伝子(1-8、19、20)のいずれかでLQTS患者の70%に見られます。 現在知られている1 3種類のLQTの中で、最も一般的なものは、それぞれ心臓イオンチャネル遺伝子、KCNQ1、KCNH2、およびSCN5Aの欠損に起因するLQTS1、LQTS2、およ すべてのLQTS原因突然変異の90%は、これらの3つの遺伝子に見られます(1-8、19、20)。 残りの1 0個の遺伝子の突然変異はまれであり、現在知られている全てのLQTS突然変異のわずか1 0%を構成する。
QT延長症候群は通常常染色体優性疾患ですが、lqts患者の5〜10%で単一遺伝子または異なる遺伝子に複数の変異が見られることがあります(21、22)。 複数の変異を有する患者は、単一の変異を有する患者と比較してより長いQTcを示すことができ、そのような患者は、生命を脅かす心臓事象のための3.5倍(21, 22)
KCNQ1遺伝子の変異は、すべてのLQTSの中で最も一般的な形態であり、LQTS1型(LQTS1)と呼ばれる。 すべてのLQTS原因突然変異の50%がKCNQ1遺伝子に見出される(20)。 Kvlqt1(Kv7.1とも呼ばれる)は、KCNQ1遺伝子によって作られたタンパク質であり、細胞内の小胞体に四量体を形成し、kcne1によってコードされるタンパク質ミンクと推定上共集合し、心筋細胞の原形質膜に輸送され、そこで心臓組織の活動電位の再分極を促進するゆっくりと活性化する電流を媒介し、この電流はIKsとして知られています(図1)。 LQTS1原因KCNQ1変異は、主にミスセンス変異であり、まれに、彼らはC末端領域(におけるフレームシフト変異である可能性があります23)。 KCNQ1変異キャリアにおける心臓不整脈は、アドレナリン作動性ドライブ、例えば、感情的ストレス、身体運動、ダイビング、水泳(24-26)によってトリガされます。 Kvlqt1の膜貫通ドメインに変異を有する患者は、LQTS関連の心臓イベントのリスクが高く、交感神経刺激に対する感受性が高い(26、27)。 ミスセンス変異を有する患者は、非センスまたは切り捨て変異を有する患者と比較してリスクが高い(27、28)。 KCNQ1遺伝子における3′-UTRの変化はまた、おそらく遺伝子の発現に影響を与えることによって、不整脈感受性に有意に影響を与える(29)。 KCNQ1突然変異による不整脈の患者はβ遮断薬にかなりよく答えますが、何人かの患者はまだこの薬物に対してより少なく敏感また更に抵抗力があ 最近の記事では、Barsheshet et al. (30)は、Kvlqt1の細胞質ループ(cループ)領域の外側に変異を有する患者は、β遮断薬に応答性が低いと主張した。
1
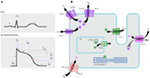
図1. 心室活動電位に寄与するイオン電流(A)および心筋細胞の模式図(のみ)遺伝性不整脈症候群(B)の病因に関与するタンパク質を表示する。 (A)では、活動電位はECGの間の行為のおおよその時間と一直線に並ぶ。 (B)では、qt延長症候群4型に関与するアダプタータンパク質であるankyrin-Bは描かれていない。
図1. 心室活動電位に寄与するイオン電流(A)および心筋細胞の模式図(のみ)遺伝性不整脈症候群(B)の病因に関与するタンパク質を表示する。 (A)では、活動電位はECGの間の行為のおおよその時間と一直線に並ぶ。 (B)では、qt延長症候群4型に関与するアダプタータンパク質であるankyrin-Bは描かれていない。
kcnq1遺伝子におけるホモ接合または複合ヘテロ接合変異は、劣性型のJervell and Lange-Nielsen症候群(JLNS)、タイプ1(31)を引き起こす可能性があります。 JLNS患者は、重度の心臓不整脈および難聴にも罹患している(31、32)。 JLNに起因するKCNQ1変異を有する患者は、通常、機能的なIkを有さない(28、31-33)。 KCNQ1にホモ接合性または複合ヘテロ接合性変異を有するにもかかわらず、一部の患者は難聴を有さない可能性があり、そのような症例は常染色体劣性LQTS1と呼ばれる(13,32)。 これらの患者では、少量の機能的IKs電流(全IKsの<10%)が依然として存在し、聴覚機能を維持する可能性がありますが、jlns患者(13、32)と同様に心臓リズム欠損
LQTS2
このタイプのLQTSはLQTS1と同様に流行しており、検出可能な変異を有するLQTS患者の35〜40%を占めています(1, 20, 24, 25). KCNH2は、急速に活性化遅延整流器K+電流(IKr)のαサブユニットであるHERGタンパク質(Kv11.1)をコードしています。 Kv11.1チャネル機能を低下させるこの遺伝子の病原性変異は、QT間隔の持続時間を延長し(図1)、LQTS2に起因する。 LQTS2における失神発作の二十から九パーセントは、休息/睡眠中に発生し、失神発作のわずか13%が運動中に発生することが報告された(25、34)。 突然の驚くべきノイズは、例えば、目覚まし時計リング、ドアベル、電話リングは、典型的には、これらの患者(におけるsyncopal攻撃をトリガ24、34)。 Kv11.1(KCNH2遺伝子によってコードされる)の孔形成領域に変異を有する患者は、非孔領域変異を有する患者と比較して不整脈関連心臓事象のリスクが高 新生児では、2:1房室(AV)ブロックが優先的にKCNH2変異(と関連付けられている36)。 LQTSによって複雑な完全なAVブロックはまた、KCNH2遺伝子(の変異を有する成人患者の17%で発見された37)。 KCNH2のホモ接合変異はまれであり、存在する場合、患者は2と、LQTSの重篤な形態に苦しみます:1AVブロックと重度の心室性不整脈,子宮内の段階の間だけでなく、出生後(11,38-40). LQTS2病理に記載されている多数の変異に加えて、KCNH2遺伝子の一般的な多型変異体はまた、疾患の重症度を調節することができる。 興味深い例は、KCNH2におけるK897T多型(SNP)であり、これは一般集団の33%に存在する(41)。 K8 9 7Tは、KCNH2突然変異の病原性を悪化させることが報告された(4 2、4 3)。
LQTS3
Nav1.図5は、電圧依存性心臓Na+チャネルの細孔形成αサブユニットであり、SCN5A遺伝子によってコードされる積分膜タンパク質であり、心臓活動電位の開始 心臓N A+チャネルは、細孔形成α−サブユニット(SCN5Aによってコードされる)および1つまたは複数の補助β−サブユニットから構成される。
lqts3は、αサブユニットの高速不活性化を破壊する機能変異の増加によって引き起こされ(図1)、SCN5A遺伝子の変異は、変異を有するすべてのlqts患者の10%(1、20、24)に記載されている。 LQTS3患者は、睡眠/休息中に心臓イベントの大部分(39%)を経験し(25)、イベントの約≥13%が運動中に発生することが報告された(25)。 いくつかの例では、単一のSCN5A変異は、同じファミリー、例えば、LQTS、BrS、またはCCD(44-47)において不整脈の2つまたは3つの異なる表現型を発揮することが示 LQTS3変異を有する男性患者は、女性患者よりもはるかに早く症状を発症する可能性がある(48)。 ヘテロ接合およびホモ接合の両方のSCN5A変異が、機能的2:1AVブロックを有するLQTS3に記載されている(4 9)。 我々は最近、患者がLQTSとCCDを組み合わせていた複数の家族において、1507_1509delqkp変異を有するイランの家族を発見した(データは示されていない)。 この変異は他の国の患者でも報告されており、これは再発性のホットスポット変異であることを示唆している。 活動電位の異なる段階の間にこのような機能喪失および機能獲得特性を有する変異も、1493delkおよび1795insd変異で報告された(44、51)。
SNPはそのキャリアに対して病原性効果を発揮することもあります。 S1103Yは、アフリカ系アメリカ人(の13%に存在するSCN5A遺伝子の一般的な変異体である52)。 この変異体を有するキャリアは、不整脈および乳児突然死症候群(SIDS)のリスクが高い(52)。
LQTS4
LQTS4は、LQTSの最初の非チャネル形式を表します。 アダプタータンパク質であるANK2の突然変異は、lqts4に寄与する細胞内カルシウム過負荷につながる(53、54)。 QT延長に加えて、この症候群の患者は、洞徐脈、発作性AF、およびCPVTを有する可能性がある(54)。 ANK2突然変異の病原性の効果は厳しいに穏健派であることができ、臨床表現は突然変異の重大度によって決まります。
LQTS5
KCNE1の変異はLQTS5と関連している(図1)(55、56)。 ヘテロ接合型KCNE1変異は、それに付随する正常な対立遺伝子に支配的な負の効果を発揮することによってIkを減少させ、不整脈のリスク増加の原因となる心臓再分極の遅延をもたらす(図1)(6)。 ホモ接合型KCNE1変異を有する患者は、JLNS(タイプ2)に苦しむ(57、58)。
D85NはKCNE1遺伝子の多型であり、0に存在する。一般人口の7-1%(41)。 西尾らの研究では、 (5 9)、D8 5N多型は、LQTS患者においてより頻繁に見出され、LQTS病理におけるリスク遺伝子型となった(潜在的にはアジア集団においてのみ)。 ヨーロッパでは、D85Nは、TdPを経験した後天性LQTS(aLQTS)患者(32人の患者のコホート)の5%で報告された(60)。
LQTS6
KCNE2遺伝子は、心臓カリウムチャネルIKrの推定βサブユニットであるミンク関連ペプチド1(Mirp1)をコードしています(図1)。 KCNE2遺伝子の変異はまた、遅延整流器カリウム電流(IKr)、LQTS6(61)の病理学的基礎の急速に活性化成分の欠陥につながる可能性があります。 聴覚/音響の刺激は目覚し時計の騒音、ドアベル等を好みます。 KCNH2変異(62)と同様に、KCNE2変異キャリアにおけるsyncopal攻撃を引き起こす可能性があります。
LQTS7
この症候群はアンデルセン–タウィル症候群(ATS)としても知られています。 ATSはまれな障害であり、時折の失神および心停止によって現れる。 ECGの特徴は穏やかなQT間隔の延長、異常なUの波、頻繁な心室のectopy、二方向の心室の頻脈(VT)、および多型VTを含んでいます。 この症候群はまた、心外膜の特徴、例えば、骨格筋周期性麻痺および発達上の問題、例えば口蓋裂、低セット耳、低身長、および四肢の発達上の欠陥を示す(63)。 臨床的に診断されたATS患者の大部分は、KCNJ2(63)に変異を有することが報告された。 KCNJ2は、内向きに整流するカリウムチャネル(IK1)の細孔形成サブユニットをコードしている(図1)(64、65)。
Lqts8
ティモシー症候群(TS)としても知られており、患者はecgsに重度のQT延長を示し、100%の症例でsyndactyly、出生時の脱毛症、小さな歯と組み合わされ、浸透性の低い心臓構造奇形、精神遅滞、自閉症、顔面の異形性特徴(66)を示す(66)。 2つのサブタイプがあります:TS1(古典的な)とTS2(まれな形)。
TS2はTS1よりも心臓学的に厳しい(66、67)。 TS2患者はまた、syndactyly(67)を欠いています。 遺伝子CACNA1CをコードするL型カルシウム電流(ICa-L)のα-1サブユニットの変異は、TS(LQTS8)の両方のフォームにつながります。 Cacna1Cが主に発現している心臓および脳では、exon-8はmRNA転写物の≥80%に見出され、exon-8Aは転写物の≥20%に存在する(66)。 エクソン-8AにおけるG406R変異はTSの古典的な形態(TS1)に原因であり、エクソン-8におけるG402SはTS2でより厳しいで報告された。 このリストに新たに追加されたのはAla1473Gly変異であり、これはさらに拡大された表現型を有するTS乳児に記載されている(68)。 すべての突然変異は親のde novoまたはmosaicのいずれかであり、ICa-Lチャネルの機能のすべての結果の利得(66-69)であった。
LQTS9およびLQTS10
CAV3またはSCN4Bの変異は、後期INaで機能の増加を生じ、LQTS3様表現型(70-74)を引き起こす。 それらは、LQTS9(CAV3突然変異に関連する)およびLQTS1 0(SCN4B突然変異に関連する)として知られている。
カベオラエはEngelmanらによってうまく記述されている。 (72)原形質膜の”小さな洞窟”として。 それらは小さなコーティングされていないピットであり、原形質膜(で重要な動的および調節イベントのサイトとして考えられている72、73)。 カベオリンはカベオラエの主要なタンパク質であり、カベオリン-3(遺伝子CAV3によってコードされる)は心筋細胞および骨格筋細胞に特異的に見られる。 いくつかの心臓イオンチャネルは、特にカベオリン-3(72、73)に富化されている心筋細胞から抽出されたカベオラエに局在することが報告されている。 さらに、β-アドレナリン受容体シグナリングカスケードの成分は、カベオラエ濃縮膜(72、73)にも存在しています。
SCN4Bは、心臓ナトリウムチャネルの補助βサブユニットであるNav Β4をコードしている。 これまでのところ、一つだけの変異(L179F)は、複数の影響を受けた家族(74)とメキシコの家族でこの遺伝子で報告されています。 この遺伝子の変異は、Nav1.5電流(74)の機能の利得をもたらすことが見出された。
LQTS11
心臓では、心臓活動電位持続時間(APD)の交感神経調節は、β-アドレナリン受容体(β-AR)活性化によって媒介され、AKAP9(Yotiao)とiksチャネルのαサブユニット(Kvlqt1) AKAP9の突然変異はLQTS11(75)を引き起こす。 現在までに、AKAP9における1つの突然変異S1 5 7 0Lのみが報告されている(7 5)。
lqts12
Α-1-シントロフィン(SNTN1)遺伝子の変異はLQTS12に原因があります。 この遺伝子の突然変異は、lqts12の病理学的基礎である心臓ナトリウムチャネル(Nav1.5)の機能の獲得につながる(76)。
LQTS13
Gタンパク質結合、内向き整流カリウムチャネルサブユニット(Kir3.4)KCNJ5遺伝子によってコードされています。 この遺伝子の機能喪失変異は、LQTS13(77)を引き起こす可能性があります。 これまでのところ、唯一の変異、G387Rは、この変異を有する九人の患者を持つ中国の家族で記載されています。 Kir3.4の原形質膜発現の低下は、患者におけるLQTSの病理として示唆された。
後天性LQTS
先天性lqtsに加えて、aLQTSとして知られるLQTSの別の変種も存在し、これはカリウム流束を減少させ、心筋の再分極能力を損なう要因および物質によ よく認識されている条件は、女性の性別、低カリウム血症、および心臓カリウムチャネル(78-80)を阻害する薬物である。 一般的に処方されている多くの薬物はまた、優先的にHERGチャネル(Kv1 1.1、KCNH2遺伝子によってコードされるタンパク質)に結合して遮断し、ALQTSを素因とす 最近、特に再分極予備が損なわれたときに、IKsの遮断が薬物誘発ALQTにも寄与する可能性があることが示された(81)。 フルオキセチンとノルフルオキセチンは、in vivoとin vitroの両方で、IKsプロパティを抑制することが判明し、マークされたLQTS(81)につながった。 多型、ミンク(遺伝子KCNE1)におけるD85N、IksおよびIKrチャネルの推定βサブユニットであるMirp1(遺伝子KCNE2)におけるT8A、Q9Eは、aLQTSを引き起こすことが報告された(82、83)。 自己免疫性LQTは、抗HERG抗体を含むIggを有する患者においても報告されている(8 4)。
QT延長症候群の三つの一般的な形態における心電図的特徴
典型的なST-T波パターンは、遺伝子型LQTS患者の大部分に存在し、LQTS1、LQTS2、および場合によってはLQTS3遺伝子型を同定するために使用することができる(85、86)。
LQTSのLQTS1型は、運動時のQT間隔を短縮することなく、広いT波と関連している(図2A)。 LQTS2は、低振幅の、しばしばbifidのT波に関連しています(図2B)。 LQTS3は、長い等電セグメントと狭いベースの背の高いT波に関連付けられています(図2C)。 先天性LQTSにおけるTdP発症の一時停止依存性は、lqts2で優勢であるが、LQTS1(87)ではほとんど存在しない、遺伝子型特異的である。

図2. LQTS1、LQTS2、およびLQTS3の患者からの心電図記録。 (A)KCNQ1変異を有する18歳の男性の12リードECG。 QT間隔は延長される(Qtc=±5 0 0m s)。 STセグメントは、広いベースと比較的大きな振幅を持っています。 伝導間隔は正常です(標準的な口径測定)。 (B)KCNH2変異を有する14歳の少女の12リードECG。 QT間隔は延長されます(QTc±520ms)。 STセグメントはリードV3でノッチされ、四肢リードでは比較的低い振幅を有する。 伝導間隔は正常です(標準的な口径測定)。 (C)SCN5A変異を有する12歳の少年の12リードECG。 QT間隔は延長されます(QTc±600ms)。 STセグメントは、大きく、鋭く、狭いT波を有する長い(ほぼ)等電セグメントを有する。 伝導間隔は正常です(標準的な口径測定)。
パターンはLQTSの特定の遺伝子型を示唆しているかもしれないが、例外が頻繁に発見されている。
遺伝子型-表現型
QT延長症候群は常染色体優性疾患である。 ヘテロ接合変異キャリア間の遺伝子型および表現型分析は、LQTS1、LQTS2、およびLQTS3患者において非常に広範囲に実施されている。 小児期には、心臓イベントのリスクは、lqts1女性よりもLQTS1男性で有意に高いが、心臓イベントのリスクの有意な性別関連の違いは、LQTS2とLQTS3患者(88-91)の間で観察されなかった。 成人期(40歳以降も)、LQTS1およびLQTS2女性は、それぞれの男性(88-91)よりも心臓イベントのリスクが有意に高い可能性があります。 一般に、心臓事象の致死率は、LQTS1およびLQTS2患者よりもLQTS3患者で優勢であると思われる(91)。 LQTSを持つ女性は、妊娠中の心臓イベントのリスクが低下しているが、リスクは非常にKCNH2遺伝子(92)の変異を持つ女性では、9ヶ月の産後期間中に増加し
小児の突然の心臓死は、心臓イオンチャネル遺伝子の突然変異(93-96)によっても引き起こされる可能性がある。 原因不明のSCDを有する子供の約28%(1〜18歳、平均年齢:12.3±3.8歳)は、LQTS原因遺伝子の突然変異のキャリアであった(97)。 SIDSでは、SCN5Aの変異が優勢であるように見えた(98、99)が、KCNQ1、KCNH2、KCNE2、およびCAV3、SCN4B、およびSCN3Bの変異も見出された(100、101)。 子宮内胎児死亡もまた、心臓イオンチャネル遺伝子の欠損によるものであることが報告された(12、102)。
LQTSの臨床管理
QT間隔を延長することが知られているすべての薬物の停止、および電解質の不均衡および/または代謝状態の沈殿の修正は、(後天性)LQTS患者 LQTSの症状はしばしばアドレナリン作動性に媒介されるため、患者の運動活動への参加の制限が一般的に推奨されます(24、25)。 LQTSの臨床療法の主力はβ遮断薬である。 Propranolol、nadololおよびmetoprololの長時間作用型の準備は通常使用され、β遮断の効力は練習の心拍数の鈍化によって査定されます(例えば、nadololおよびmetoprolol>20%) (24, 25). すべてのβ遮断薬の中で、プロプラノロールとナドロールは、症状のある患者(103)でメトプロロールよりも優れていると考えられています。 さらに、β遮断は、SCDを減少させるための無声突然変異キャリアにおける予防的処置としても使用することができる(2 4)。 Barsheshetらによる研究では。 (30)、KCNQ1遺伝子のcループミスセンス変異を有する患者は、生命を脅かす心臓事象のリスクが高く、β遮断薬による治療から有意な利益を有していた。 Lqts2を持つ女性は9ヶ月の産後の期間の間に高められた危険があるので、β遮断薬はこの危険度が高い期間(の間に心臓でき事を減らすために規定さ 植込み型除細動器(ICDs)はβ遮断薬療法にもかかわらず再発失神の患者または心停止のための危険度が高いの患者のために考慮することができます(例 lqts2およびlqts3は、文書化されたQtc延長を伴う)。 ICDが禁忌または拒否され、β遮断薬が有効でない、許容されない、受け入れられない、または禁忌である高リスクLQTS患者には、左心臓交感神経脱神経(LCSD)が推奨 JLNS患者は通常、QTc>500msを有し、またリスクが高く、icdによる早期治療が推奨された患者ではβ遮断薬の有効性が不十分である(18、31)。
: サウジの視点
サウジアラビアからのLQTに関する最初の報告書は、1993年にリヤド軍病院から出版された(105)。 単一の家族からの再発発作の歴史を持つ6と48ヶ月の間の四つの幼児と幼児は、LQTS(105)と診断されました。 家族歴は、他の二人の大家族が意識の突然の喪失の同様のエピソードを持っていたし、三家族のメンバーが突然死亡したことを明らかにした(105)。 すべての症例において、初期診断はてんかんであった(105)。 数年後、2:1AVブロックと組み合わせた新生児LQTの比較的厳しい変異体を有する2つの散発的な症例報告が報告されている(106、107)。 これらの発表された臨床報告はすべて、それらのLQTの病態生理を説明することができる遺伝的所見なしであった(105-107)。
私たちは、サウジアラビアの同様の患者におけるlqtsの病理学的根拠として遺伝的欠陥を初めて報告しました。 失神の歴史と胎児、新生児および子供(12-14)の突然の原因不明の死亡を有する六つのサウジアラビアの家族を調査しました。 常染色体劣性LQTS1は、二つの家族からの子供で診断されました(図3). 常染色体劣性LQTS2は、二つの家族で診断されました(図4). ある家族では、女性患者は常染色体優性LQTS2と診断された(図5)、患者はKCNH2変異キャリアの女性で非常に一般的である病院で産後の回復期間中に失 すべての患者において、LQTS原因心臓イオンチャネル遺伝子における病原性変異の同定は、私たちに言及される前にてんかん痙攣と誤診された確認された臨床診断につながった(13、14)最近、Shinwari et al. (15)キングファイサル専門病院と研究センターからLQTS1因果KCNQ1変異、H258P、12の影響を受けた個人を持つ大家族で報告しました。 二つのキャリアだけが症候性であり、そのQTcは>500msであり、β遮断薬はある患者の臨床症状を抑制し、第二の症候性患者はICDを必要とした。
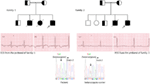
図3. 家族の血統図-1と2常染色体劣性LQTS1では、発端者は矢印で示されています。 二つのファミリーの発端者からのecgは、それぞれ557と529msのQTcで、中央に示されています。 イントロン変異は、KCNQ1遺伝子におけるc.387-5T>Aが患者に見出された(下に示されている)。 変異は矢印で示された。 塗りつぶされていない円と正方形は、突然変異の非キャリアです。 影響を受けた個体は、塗りつぶされた円(女性)と正方形(男性)として表示されます。 半分に満たされた正方形と円は、ヘテロ接合体変異を有する個体である。 死亡した個体はスラッシュで示され、発端者は矢印で示され、近親婚は=エクソン−イントロン境界で示され、エクソンに向かって矢印を指す点線で示されている。
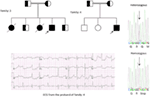
図4. 家族の血統図-常染色体劣性LQTS2を有する3および4。 家族4からの発端者のECGは、洞性頻脈、ほぼ完全なAVブロック、非常に長いQTc間隔(QT>600ms)を有する広い複雑な脱出リズムを示す血統図の下に示されている。 Kcnh2遺伝子の変異、c.3208C>T(p.Q1070X)が、矢印でマークされた患者(右に示されている)で発見された。
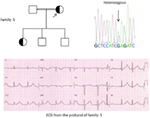
図5. 左上:家族の血統-5。 影響を受けた個体は、塗りつぶされた円(女性)で示されている発端者は、矢印で示されています。 12-発端者のリードECG(I:2)。 KCNH2遺伝子のスクリーニングは、”A”に対するヌクレオチド”G”の置換(c.2362G>A、矢印がマークされている)を示し、アミノ酸置換、P.E788Kをもたらす。.
サウジアラビアからの我々の研究(12-14)における遺伝的および臨床的知見は、いくつかの理由のために非常に興味深いです:(1)合計で、我々は六つの家族を調; (2)LQTSのすべての変異は、これらのアラブの家族でのみ報告され、新規であった;(3)変異のホモ接合性または化合物ヘテロ接合性のために、臨床表現型は、我々の研究された家族(12-14)でも重度であった。 我々は、遺伝的および表現型の観察は、サウジアラビア(108、109)における同族結婚の極端な高い割合に由来することを示唆した。 我々の研究は、サウジアラビア(12-14)の小児および青年における原因不明の心臓不整脈およびSCDsにおける中枢的な役割を発揮する際の血縁の役割に また、Kcnq1変異c.387-5T>A(NM_000218)(図3)は、同族結婚の発生率が高いため、いくつかの世代の間に共通の祖先からサウジアラビアのアシール州に広がっていたこと これまでのところ、これはサウジアラビアの人口における最も一般的なLQTS1原因突然変異であり、King Faisal Specialist HospitalとKhamis Mashayt Military Hospital(未発表)でも観察されました。 同様の結果は、LQTS2原因突然変異、c.3208C>Tについても得られた(p.Q1070X)(図4)Kcnh2遺伝子(12,14)は、サウジアラビアの創始者変異でもあり、アシール地域の多くの世代の間に分離されています(地図、図6参照)。 我々は、言及された祖先/創始者の突然変異(KCNQ1とKCNH2および他の遺伝子の両方)を持つかなりの数の個体がこの地域およびリヤド、ジェッダ、ダンマームのような大都市にも都市移住のために存在することを予測するだろう。 Lqtsに病原性のあるより多くの創始者または先祖の突然変異は、同族結婚の割合が高いため、サウジアラビアの他の州では非常に想定されています。
図6. サウジアラビアの地図(礼儀:ウィキペディア)。 Assir領域は太い線でマークされており、家族1-4に記載されているKCNQ1およびKCNH2遺伝子の創始者変異を発見しました。
LQTSは常染色体優性疾患であるため、主にヘテロ接合変異キャリア患者を観察することが期待されています。 しかし、我々の研究では、主に劣性突然変異(12-14)を有する患者を同定した。 明らかに、これらの患者は最初に症候性になり、主に王子スルタン心臓センターと呼ばれ、私たちの注意の下でそれらをもたらしました。 それにもかかわらず、我々の調査からの知見は、劣性LQTが小児において致命的な臨床表現型を有する可能性があり、サウジアラビアでは珍しいことでは ヘテロ接合変異キャリアは不整脈やその合併症を発症しやすいため、地元の一般医師、心臓病専門医、臨床遺伝学者を共通のプラットフォームに連れて、リスクのある個人を特定するための協調的なイニシアチブが取られるべきである。 さらに、現時点では、他の家族性不整脈障害、例えば、CPVT、SQTS、BrS、AFなどの遺伝情報も持っていません。 専門の心臓遺伝学センターは、遺伝的欠陥、突然変異を検索し、遺伝性不整脈のすべての形態で遺伝子型表現型研究を行うためのイニシアチブを取る また、多くの場合、不整脈原因突然変異は起源がデノボであるため、すべての遺伝的不整脈が家族歴を有するわけではないこと、すなわち発端者がその家族の最初の患者であり、彼/彼女が下流世代に突然変異を伝達する源であることを考慮しておくべきである(110、111)。 サウジアラビアでは近親婚の割合が非常に高いため、この国では先天性不整脈に重要な役割を果たす多くの創設者変異が期待されています。 これらの創設者の突然変異を特定することは、この国で効果的な婚前および前症候性遺伝カウンセリングを開発する上で私たちを容易にする私た LQTS原因遺伝子の変異は、臨床浸透度の変動を与えることができ、一つの極端に、いくつかのキャリアは、おそらく完全に健康である可能性がありますが、 幼児または大人の突然死は家族の大量の心理的で、感情的な重荷を提起します、キャリアのスクリーニングは不整脈およびSCDsの致命的な結果からキャリアを非常に効果的に防ぐことができる簡単な薬物、例えば、β遮断薬(また行動の修正)があるので必要です。 LQTSの原因遺伝子のhomozygous突然変異の個人は胎児のbrady-tachyarrhythmiasを含む死亡率の厳しい罹患率そして極度で高い率があり、多くの場合流産、妊娠した母の自発の中絶(12-14)。
サウジアラビアでは、不整脈関連遺伝子およびそれらを調節する遺伝子における様々なSnpの有病率についてもあまり研究が行われていない。 Splawski et al. (112)アフリカ系アメリカ人の不整脈に関連付けられているSCN5A遺伝子の一般的なS1103Y変異体を説明しました。 Y1103と呼ばれる変異対立遺伝子は、それによってアフリカ系(52、112)の人の心臓不整脈の確率を高める、チャネル活性化を加速するための責任があります。 K393Nは、米国のLQTS1患者で報告されたKCNQ1遺伝子の変異体であるが、アラブ集団では、この変異体を個人の2%で検出している(未公開データ)。 アラブ人のKCNQ1遺伝子のK393N変異体が、アフリカ系アメリカ人のS1103Y(SCN5A)変異体または日本人集団のD85N(KCNE1)変異体に匹敵するかどうかも調査に値する可能性がある(59)。
利益相反に関する声明
著者らは、本研究は、利益相反の可能性があると解釈される可能性のある商業的または財政的関係がない場合に行われた
謝辞
Arthur Wilde教授、Connie Bezzina教授、およびジャーナル”Heart”の出版社に、この原稿の図1を使用する許可を与えてくれたことに心から感謝しています。 (113).
1. ブイヤン-ザ 遺伝性心臓不整脈症候群の臨床的および遺伝的スペクトル。 アムステルダム:アムステルダム大学(2009)。
2. Priori SG,Aliot E,Blømstrom-Lundqvist C,Bossaert L,Breithardt G,Brugada P,et al. 心臓突然死に関するタスクフォース、心臓病のヨーロッパの社会。 2002年4月3日から18日まで放送された。 土井:10.1053/2001.0214
クロスリファレンス全文
3. Wang Q,Shen J,Splawski I,Atkinson D,Li Z,Robinson JL,et al. 遺伝性心臓不整脈、QT延長症候群に関連するSCN5A変異。 Cell(1 9 9 5)8 0:8 0 5−1 1. 土井:10.1016/0092-8674(95)90359-3
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
4. カランME,Splawski I,Timothy KW,Vincent GM,Green ED,Keating MT. 心臓不整脈の分子基盤:HERG変異はQT延長症候群を引き起こす。 Cell(1 9 9 5)8 0:7 9 5−8 0 3. 土井:10.1016/0092-8674(95)90358-5
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
5. Wang Q,Curran ME,Splawski I,Burn TC,Millholland JM,VanRaay TJ,et al. 新規カリウムチャネル遺伝子の位置クローニング:KVLQT1変異は、心臓不整脈を引き起こす。 Nat Genet(1 9 9 6)1 2:1 7−2 3. doi:10.1038/ng0196-17
Pubmed要約/Pubmed全文|CrossRef全文
6. Splawski I,Tristani-Firouzi M,Lehmann MH,Sanguinetti MC,Keating MT. Hmink遺伝子の変異はqt延長症候群を引き起こし,Iks機能を抑制する。 Nat Genet(1 9 9 7)1 7:3 3 8−4 0. doi:10.1038/ng1197-338
Pubmed要約/Pubmed全文|CrossRef全文
7. サンギネッティMC、江C、カランME、キーティングMT。 遺伝性不整脈と後天性不整脈との間の機械的リンク:HERGはIKrカリウムチャネルをコードする。 Cell(1 9 9 5)8 1:2 9 9−3 0 7. 土井:10.1016/0092-8674(95)90340-2
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
8. Neyroud N,Tesson F,Denjoy I,Leibovici M,Donger C,Barhanin J,et al. カリウムチャネル遺伝子KVLQT1の新規変異は、Jervellとランゲ-ニールセン心auditory症候群を引き起こします。 Nat Genet(1 9 9 7)1 5:1 8 6−9. doi:10.1038/ng0297-186
Pubmed要約/Pubmed全文|CrossRef全文
9. Kucera JP,Rohr S,Rudy Y.インターカレートディスクにおけるナトリウムチャネルの局在は、心臓伝導を調節する。 Circ Res(2 0 0 2)9 1:1 1 7 6−8 2. 土井:10.1161/01.RES.0000046237.54156.0A
Pubmed要約/Pubmed全文|CrossRef全文
10. Oxford E M,Musa H,Maass K,Coombs W,Taffet SM,Delmar M. Circ Res(2 0 0 7)1 0 1:7 0 3−1 1. 土井:10.1161/107.154252
Pubmed要約/Pubmed全文|CrossRef全文
11. Rohr S.インターカレートディスクにおける機械的および電気的接合間の分子クロストーク。 Circ Res(2 0 0 7)1 0 1:6 3 7−9. 土井:10.1161/107.161901
クロスリファレンス全文
12. Bhuiyan ZA,Momenah TS,Gong Q,Amin AS,Ghamdi SA,Carvalho JS,et al. HERGのほぼ不在による再発子宮内胎児の損失:ホモ接合ナンセンスHERG Q1070X変異の臨床的および機能的特徴付け。 心のリズム(2008)5:553-61. 土井:10.1016/j.hrthm.2008.01.020
Pubmed要約/Pubmed全文|CrossRef全文
13. Bhuiyan ZA,Momenah TS,Amin TS,Al-Khadra AS,Alders M,Wilde AAM,et al. Kcnq1におけるエクソン-2の不完全なスキップにつながるイントロン変異は、ジャーベルとランゲ-ニールセン症候群の聴覚を救出します。 Prog Biophys Mol Biol(2 0 0 8)9 8:3 1 9−2 7. 土井:10.1016/j.pbiomolbio.2008.10.004
Pubmed要約/Pubmed全文|CrossRef全文
14. Bhuiyan ZA,Al-Shahrani S,Al-Khadra AS,Al-Ghamdi S,Al-Kalaf K,Mannens MMAM,et al. サウジアラビアの6家族の小児におけるQT延長症候群の臨床および遺伝子解析:彼らは異なっていますか? Pediatr Cardiol(2009)30:490-501. ドイ:10.1007/s00246-008-9377-y
Pubmed要約/Pubmed全文|CrossRef全文
15. Shinwari ZM,Al-Hazzani A,Dzimiri N,Tulbah S,Mallawi Y,Al-Fayyadh M,et al. QT延長症候群を有する大規模なサウジ家族における新規KCNQ1変異の同定:臨床的結果と予防的影響。 Clin Genet(2 0 1 3)8 3:3 7 0−4. ドイ:10.1111/j.1399-0004.2012.01914.x
Pubmed要約/Pubmed全文|CrossRef全文
16. Schwartz PJ,Stramba−Badiale M,Crotti L,Pedrazzini M,Besana A,Bosi G,et a l. 先天性QT延長症候群の有病率。 循環(2009)120:1761-7. 土井:10.1161/109.863209
Pubmed要約/Pubmed全文|CrossRef全文
17. シュワルツPJ、モスAJ、ヴィンセントGM、クランプトンRS。 QT延長症候群の診断基準。 更新しました。 Circulation(1 9 9 3)8 8:7 8 2−4. 土井:10.1161/01.CIR88.2.782
クロスリファレンス全文
18. Priori SG,Wilde AA,Horie M,Cho Y,Behr ER,Berul C,et al. エグゼクティブサマリー:遺伝性原発性不整脈症候群を有する患者の診断および管理に関するHRS/EHRA/APHRSコンセンサス声明。 Europace(2013)15:1389-406. 土井:10.1093/欧州/eut272
クロスリファレンス全文
19. Liu JF,Jons C,Moss AJ,McNitt S,Peterson DR,Qi M,et al. シンドロームの登録簿。 Qt延長症候群の小児および青年における再発性失神およびその後の致命的または致命的に近い事象の危険因子。 J Am Coll Cardiol(2011)57:941-50. 土井:10.1016/j.jacc.2010.10.025
Pubmed要約/Pubmed全文|CrossRef全文
20. Splawski I,Shen J,Timothy KW,Lehmann MH,Priori S,Robinson JL,et al. QT延長症候群遺伝子における突然変異のスペクトル。 KVLQT1、HERG、SCN5A、KCNE1、およびKCNE2。 循環(2000)102:1178-85。 土井:10.1161/01.CIR102.10.1178
Pubmed要約|Pubmed全文|CrossRef全文
21. Westenskow P,Splawski I,Timothy KW,Keating MT,Sanguinetti MC. 複合変異:重度のQT延長症候群の一般的な原因。 循環(2004)109:1834-41. 土井:10.1161/01.CIR0000125524.34234.13
Pubmed要約/Pubmed全文|CrossRef全文
22. Mullally J,Goldenberg I,Moss AJ,Lopes CM,Ackerman MJ,Zareba W,et al. QT延長症候群および複数の変異を有する患者の生命を脅かす心臓事象のリスク。 心のリズム(2013)10:378-82. 土井:10.1016/j.hrthm.2012.11.006
Pubmed要約/Pubmed全文|CrossRef全文
23. Napolitano C,Priori SG,Schwartz PJ,Bloise R,Ronchetti E,Nastoli J,et al. QT延長症候群における遺伝子検査:臨床診療におけるジェノタイピングへの効率的なアプローチの開発と検証。 JAMA(2005)294:2975-80. 土井:10.1001/jama.294.23.2975
Pubmed要約/Pubmed全文|CrossRef全文
24. ローデンDM. 臨床実習。 QT延長症候群。 N Engl J Med(2 0 0 8)3 5 8:1 6 9−7 6. doi:10.1056/Nejmcp0706513
CrossRef全文
25. Schwartz PJ,Priori SG,Spazzolini C,Moss AJ,Vincent GM,Napolitano C,et al. Qt延長症候群における遺伝子型-表現型相関:生命を脅かす不整脈の遺伝子特異的トリガー。 循環(2001)103:89-95. 土井:10.1161/01.CIR103.1.89
クロスリファレンス全文
26. Ackerman MJ、テスター DJ、ポーター CJ。 水泳、遺伝性qt延長症候群のための遺伝子特異的不整脈誘発トリガー。 Mayo Clin Proc(1 9 9 9)7 4:1 0 8 8−9 4. 土井:10.4065/74.11.1088
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
27. Kapa S,Tester DJ,Salisbury BA,Harris-Kerr C,Pungliya MS,Alders M,et al. QT延長症候群の遺伝子検査:病原性変異と良性変異を区別する。 循環(2009)120:1752-60。 土井:10.1161/109.863076
Pubmed要約/Pubmed全文|CrossRef全文
28. Moss AJ,Shimizu W,Wilde AA,Towbin JA,Zareba W,Robinson JL,et al. KCNQ1遺伝子を含む突然変異の位置、コードタイプ、および生物物理学的機能による1型QT延長症候群の臨床的側面。 循環(2007)115:2481-9. 土井:10.1161/106.665406
Pubmed要約/Pubmed全文|CrossRef全文
29. Amin AS,Giudicessi JR,Tijsen AJ,Spanjaart AM,Reckman YJ,Klemens CA,et al. Kcnq1エンコードKv7.1カリウムチャネルの3’非翻訳領域の変異体は、対立遺伝子特異的な方法で1型QT延長症候群の患者の疾患の重症度を変更します。 Eur H Eart J(2 0 1 2)3 3:7 1 4−2 3. doi:10.1093/eurheartj/ehr473
Pubmed要約/Pubmed全文|CrossRef全文
30. Barsheshet A,Goldenberg I,O-Uchi J,Moss AJ,Jons C,Shimizu W,et al. KCNQ1チャネルの細胞質ループにおける変異および生命を脅かす事象のリスク: タイプ1QT延長症候群におけるβ遮断薬療法に対する突然変異特異的応答のための含意。 循環(2012)125:1988-96。 土井:10.1161/111.048041
Pubmed要約/Pubmed全文|CrossRef全文
31. Schwartz PJ,Spazzolini C,Crotti L,Bathen J,Amlie JP,Timothy K,et al. JervellおよびLange-Nielsen症候群:自然史、分子基礎、および臨床転帰。 循環(2006)113:783-90. 土井:10.1161/105.592899
Pubmed要約/Pubmed全文|CrossRef全文
32. ブイヤン-ザ-ワイルド-アア… 心臓と聴覚のIKsは、耳は心臓よりも少ないで行うことができます。 2013年(平成25年)6月14日-3月3日に放送された。 土井:10.1161/CIRCGENETICS.113.000143
クロスリファレンス全文
33. Wollnik B、Schroeder BC、Kubisch C、Esperer HD、Wieacker P、Jentsch TJ。 遺伝性心臓不整脈で見られる優性および劣性KVLQT1K+チャネル変異の病態生理学的メカニズム。 Hum Mol Genet(1997)6:1943-9. ドイ:10.1093/hmg/6.11.1943
クロスリファレンス全文
34. Wilde AA,Jongbloed RJ,Doevendans PA,Düren DR,Hauer RN,van Langen IM,et al. 不整脈イベントのトリガーとしての聴覚刺激は、KVLQT1関連患者(LQTS1)からHERG関連(LQTS2)患者を区別します。 J A M Coll Cardiol(1 9 9 9)3 3:3 2 7−3 2. ドイ:10.1016/S0735-1097(98)00578-6
CrossRef全文
35. Moss AJ,Zareba W,Kaufman ES,Gartman E,Peterson DR,Benhorin J,et al. ヒトether-a-go-go関連遺伝子カリウムチャネルの細孔領域に変異を伴うQT延長症候群における不整脈事象のリスクの増加。 循環(2002)105:794-9. doi:10.1161/hc0702.105124
Pubmed要約|Pubmed全文|CrossRef全文
36. Lupoglazoff JM,Denjoy I,Villain E,Fressart V,Simon F,Bozio A,et al. 新生児におけるqt延長症候群:HERG変異およびkcnq1変異を伴う洞徐脈に関連する伝導障害。 J Am Coll Cardiol(2004)43:826-30. 土井:10.1016/j.jacc.2003.09.049
Pubmed要約/Pubmed全文|CrossRef全文
37. Chevalier P,Bellocq C,Millat G,Piqueras E,Potet F,Schott JJ,et al. 房室ブロックを複雑にするTorsades de pointes:遺伝的素因の証拠。 心のリズム(2007)4:170-4. 土井:10.1016/j.hrthm.2006.10.004
Pubmed要約/Pubmed全文|CrossRef全文
38. Hoorntje T,Alders M,van Tintelen P,van der Lip K,Sreeram N,van der Wal A,et al. HERGタンパク質のホモ接合早期切断:ヒトHERGノックアウト。 Circulation(1 9 9 9)1 0 0:1 2 6 4−7. 土井:10.1161/01.CIR100.12.1264
Pubmed要約|Pubmed全文|CrossRef全文
39. Piippo K,Laitinen P,Swan H,Toivonen L,Viitasalo M,Pasternack M,et al. HERGカリウムチャネル変異のホモ接合性は、Qt延長症候群の重度の形態を引き起こす:フィンランド人の見かけの創始者変異の同定。 J Am Coll Cardiol(2000)35:1919-25. ドイ:10.1016/S0735-1097(00)00636-7
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
40. Johnson WH Jr,Yang P,Yang T,Lau YR,Mostella BA,Wolff DJ,et al. 重度の新生児QT延長症候群を引き起こすホモ接合性HERG変異の臨床的、遺伝的、および生物物理学的特性評価。 Pediatr Res(2 0 0 3)5 3:7 4 4−8. 土井:10.1203/01.PDRだ0000059750.17002.B6
Pubmed要約/Pubmed全文|CrossRef全文
41. アッカーマンMJ、テスター DJ、ジョーンズGS、ウィルML、バロー CR、カラン私。 心臓カリウムチャネル変異体の民族的差異: 先天性qt延長症候群の突然の心臓死および遺伝子検査に対する遺伝的感受性に対する影響。 Mayo Clin Proc(2 0 0 3)7 8:1 4 7 9−8 7. 土井:10.4065/78.12.1479
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
42. Crotti L,Lundquist AL,Insolia R,Pedrazzini M,Ferrandi C,De Ferrari GM,et al. KCNH2-K897Tは潜在的な生来の長いQTシンドロームの遺伝の修飾語です。 循環(2005)112:1251-8. 土井:10.1161/105.549071
Pubmed要約/Pubmed全文|CrossRef全文
43. Nof E,Cordeiro JM,Pérez GJ,Scornik FS,Calloe K,Love B,et al. 一般的な一塩基多型は、幼児突然死につながるQT長型2症候群を悪化させることができます。 (2010)3:199-206. 土井:10.1161/CIRCGENETICS.109.898569
Pubmed要約|Pubmed全文|CrossRef全文
44. Bezzina C,Veldkamp MW,van Den Berg MP,Postma AV,Rook MB,Viersma JW,et al. 長いQTとBrugada症候群の両方を引き起こす単一のNa(+)チャネル変異。 Circ Res(1 9 9 9)8 5:1 2 0 6−1 3. 土井:10.1161/01.RES.85.12.1206
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
45. Kyndt F,Probst V,Potet F,Demolombe S,Chevallier JC,Baro I,et al. 大規模なフランスの家族で孤立した心臓伝導欠損またはBrugada症候群のいずれかにつながる新規SCN5A変異。 循環(2001)104:3081-6. doi:10.1161/hc5001.100834
Pubmed要約|Pubmed全文|CrossRef全文
46. Makita N,Behr E,Shimizu W,Horie M,Sunami A,Crotti L,et al. SCN5AにおけるE1784K変異は、3型QT延長症候群の混合臨床表現型に関連付けられています。 J Clin Invest(2008)118:2219-29. doi:10.1172/JCI34057
Pubmed要約/Pubmed全文|CrossRef全文
47. Keller DI,Acharfi S,Delacrétaz E,Benammar N,Rotter M,Pfammatter JP,et al. ナトリウムチャネル不活性化におけるQ1507残基の役割:SCN5A、delQKP1507-1509、長いQT症候群を引き起こしているの新規変異。 J Mol Cell Cardiol(2 0 0 3)3 5:1 5 1 3−2 1. 土井:10.1016/j.yjmcc.2003.08.007
Pubmed要約/Pubmed全文|CrossRef全文
48. Priori SG,Schwartz PJ,Napolitano C,Bloise R,Ronchetti E,Grillo M,et al. QT延長症候群におけるリスク層別化。 N Engl J Med(2 0 0 3)3 4 8:1 8 6 6−7 4. doi:10.1056/Nejmoa022147
Pubmed要約/Pubmed全文|CrossRef全文
49. Lupoglazoff JM,Cheav T,Baroudi G,Berthet M,Denjoy I,Cauchemez B,et al. 機能的な二対一房室ブロックを有するQT延長症候群におけるホモ接合性SCN5A変異。 Circ Res(2001)89:E16–21. 土井:10.1161/hh1401.095087
CrossRef全文
50. 市R、張Y、ヤンC、黄C、周X、強H、ら。 心臓ナトリウムチャネル変異delQKP1507-1509は、lqt3、伝導障害、拡張型心筋症、および若者の突然死の高い発生率の拡大表現型スペクトルに関連付けられています。 Europace(2008)10:1329-35. doi:10.1093/europace/eun202
Pubmed要約/Pubmed全文|CrossRef全文
51. Zumhagen S,Veldkamp MW,Stallmeyer B,Baartscheer A,Eckardt L,Paul M,et al. 喪失および機能獲得特性を有する心臓ナトリウムチャネル遺伝子SCN5Aにおけるヘテロ接合欠失変異は、BrugadaまたはQT延長症候群の徴候なしに、単離された PLOS One(2 0 1 3)8:e6 7 9 6 3. doi:10.1371/ジャーナル。ポネ0067963
Pubmed要約/Pubmed全文|CrossRef全文
52. Plant LD,Bowers PN,Liu Q,Morgan T,Zhang T,State MW,et al. アフリカ系アメリカ人における突然の乳児死亡に関連する一般的な心臓ナトリウムチャネル変異体、SCN5A S1103Y.J Clin Invest(2006)116:430-5。 土井:10.1172/JCI25618
Pubmed要約/Pubmed全文|CrossRef全文
53. Mohler PJ,Schott JJ,Gramolini AO,Dilly KW,Guatimosim S,duBell WH,et al. Ankyrin-Bの突然変異によりタイプ4の長いQTの心臓不整脈および突然の心臓死を引き起こします。 Nature(2 0 0 3)4 2 1:6 3 4−9. doi:10.1038/nature01335
Pubmed要約/Pubmed全文|CrossRef全文
54. Schott JJ,Charpentier F,Peltier S,Foley P,Drouin E,Bouhour JB,et al. Qt延長症候群の遺伝子の染色体4q25-27へのマッピング。 A M J Hum Genet(1 9 9 5)5 7:1 1 1 4−2 2.
Pubmed要約|Pubmed全文
55. Barhanin J、Lesage F、Guillemare E、Fink M、Lazdunski M、Romey G.K(V)LQT1およびlsK(minK)タンパク質は、i(Ks)心臓カリウム電流を形成するために会合する。 Nature(1 9 9 6)3 8 4:7 8−8 0。 doi:10.1038/384078a0
Pubmed要約/Pubmed全文|CrossRef全文
56. Sanguinetti MC,Curran ME,Zou A,Shen J,Spector PS,Atkinson DL,et al. K(V)LQT1とミンク(IsK)タンパク質のCoassemblyは、心臓I(Ks)カリウムチャネルを形成する。 Nature(1 9 9 6)3 8 4:8 0−3. 土井:10.1038/384080a0
Pubmed要約/Pubmed全文|CrossRef全文
57. Schulze-Bahr E,Wang Q,Wedekind H,Haverkamp W,Chen Q,Sun Y,et al. KCNE1変異は、JervellおよびLange-Nielsen症候群を引き起こす。 Nat Genet(1 9 9 7)1 7:2 6 7−8. doi:10.1038/ng1197-267
CrossRef全文
58. Duggal P、Vesely MR、Wattanasirichaigoon D、Villafane J、Kaushik V、Beggs AH。 Qt延長症候群のJervellおよびLange-NielsenおよびRomano-Wardの両方の形態に関連するIKsの遺伝子の変異。 Circulation(1 9 9 8)9 7:1 4 2−6. 土井:10.1186/1471-2350-9-24
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
59. 西尾Y,牧山T,伊藤H,坂口T,大野S,Gong YZ,et al. D85N、KCNE1多型は、qt延長症候群の疾患を引き起こす遺伝子変異体です。 J Am Coll Cardiol(2009)54:812-9. 土井:10.1016/j.jacc.2009.06.005
Pubmed要約/Pubmed全文|CrossRef全文
60. Paulussen AD,Gilissen RA,Armstrong M,Doevendans PA,Verhasselt P,Smeets HJ,et al. 薬物誘発性qt延長症候群患者におけるKCNQ1、KCNH2、SCN5A、KCNE1、およびKCNE2の遺伝的変異。 Mol Med(2 0 0 4)8 2:1 8 2−8. ドイ:10.1007/s00109-003-0522-z
Pubmed要約/Pubmed全文|CrossRef全文
61. Abbott GW,Sesti F,Splawski I,Buck ME,Lehmann MH,Timothy KW,et al. Mirp1はHERGとIKrカリウムチャネルを形成し、心臓不整脈と関連している。 Cell(1 9 9 9)9 7:1 7 5−8 7. doi:10.1016/S0092-8674(00)80728-X
Pubmed要約/Pubmed全文|CrossRef全文
62. Gordon E、Panaghie G、Deng L、Bee KJ、Roepke TK、Krogh−Madsen T、et a l. 聴覚刺激および血清電解質不均衡によって誘発される心臓不整脈を有する患者におけるKCNE2変異。 Cardiovasc Res(2008)77:98-106. doi:10.1093/cvr/cvm030
Pubmed要約/Pubmed全文|CrossRef全文
63. プラスター NM,Tawil R,Tristani-Firouzi M,Canún S,Bendahhou S,角田A,et al. Kir2.1の突然変異は、アンデルセン症候群の発達およびエピソード的な電気表現型を引き起こす。 Cell(2 0 0 1)1 0 5:5 1 1−9. 土井:10.1016/0092-8674(01)00342-7
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
64. 久保Y,ボールドウィンTJ,ヤンYN,ヤンLY. マウス内側整流器カリウムチャネルの一次構造と機能発現。 Nature(1 9 9 3)3 6 2:1 2 7−3 3. doi:10.1038/362127a0
Pubmed要約/Pubmed全文|CrossRef全文
65. Raab-Graham KF,Radeke CM,Vandenberg CA. 分子クローニングと人間の心臓内側整流器カリウムチャネルの発現。 Neuroreport(1994)5:2501-5. 土井:10.1097/00001756-199412000-00024
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
66. Splawski I,Timothy KW,Sharpe LM,Decher N,Kumar P,Bloise R,et al. Ca(V)1.2カルシウムチャネル機能不全は、不整脈や自閉症を含む多系統障害を引き起こします。 セル(2004)119:19-31. 土井:10.1016/j。2004.09.011
Pubmed要約/Pubmed全文|CrossRef全文
67. Splawski I,Timothy KW,Decher N,Kumar P,Sachse FB,Beggs AH,et al. 心臓L型カルシウムチャネル変異によって引き起こされる重度の不整脈障害。 Proc Natl Acad Sci U S A(2 0 0 5)1 0 2:8 0 8 9−9 6. ドイ:10.1073/pnas.0502506102
Pubmed要約/Pubmed全文|CrossRef全文
68. Gillis J,Burashnikov E,Antzelevitch C,Blaser S,Gross G,Turner L,et al. QT延長、syndactyly、共同拘縮、打撃および新しいCACNA1Cの突然変異:ティモシーシンドロームのスペクトルを拡大すること。 A m J Med Genet A(2 0 1 2)1 5 8A:1 8 2−7. 土井:10.1002/ajmg.a.34355
Pubmed要約/Pubmed全文|CrossRef全文
69. Dufendach KA,Giudicessi JR,Boczek NJ,Ackerman MJ. 母親のモザイク主義は、1型ティモシー症候群の新生児診断を混乱させる。 Pediatrics(2013)131:e1991–5. ドイ:10.1542/peds.2012-2941
Pubmed要約/Pubmed全文|CrossRef全文
70. Vatta M,Ackerman MJ,Ye B,Makielski JC,Ughanze EE,Taylor EW,et al. 変異体カベオリン-3は、永続的な後期ナトリウム電流を誘導し、QT延長症候群に関連付けられています。 流通(2006年)114:2104–12. 土井:10.1161/106.635268
Pubmed要約/Pubmed全文|CrossRef全文
71. Cronk LB,Ye B,Kaku T,Tester DJ,Vatta M,Makielski JC,et al. 乳幼児突然死症候群のための新しいメカニズム:カベオリン-3の変異に続発する持続的な後期ナトリウム電流。 心のリズム(2007)4:161-6. 土井:10.1016/j.hrthm.2006.11.030
Pubmed要約/Pubmed全文|CrossRef全文
72. Engelman JA,Zhang X,Galbiati F,Volonte D,Sotgia F,Pestell RG,et al. カベオリン遺伝子ファミリーの分子遺伝学:ヒト癌、糖尿病、アルツハイマー病、および筋ジストロフィーへの影響。 A M J H Um Genet(1 9 9 8)6 3:1 5 7 8−8 7. 土井:10.1086/302172
CrossRef全文
73. Balijepalli RC,Foell JD,Hall DD,Hell JW,Kamp TJ. Caveolar高分子シグナリング複合体への心臓L型Ca(2+)チャネルの局在は、ベータ(2)-アドレナリン作動性調節のために必要です。 Proc Natl Acad Sci U S A(2 0 0 6)1 0 3:7 5 0 0−5. ドイ:10.1073/pnas.0503465103
Pubmed要約/Pubmed全文|CrossRef全文
74. Medeiros-Domingo A,Kaku T,Tester DJ,Iturralde-Torres P,Itty A,Ye B,et al. 先天性QT延長症候群におけるSCN4Bエンコードされたナトリウムチャネルbeta4サブユニット。 循環(2007)116:134-42. 土井:10.1161/106.659086
Pubmed要約/Pubmed全文|CrossRef全文
75. Chen L,Marquardt ML,Tester DJ,Sampson KJ,Ackerman MJ,Kass RS. A-キナーゼアンカリング蛋白質の変異は、QT延長症候群を引き起こす。 Proc Natl Acad Sci U S A(2 0 0 7)1 0 4:2 0 9 9 0−5. ドイ:10.1073/pnas.0710527105
Pubmed要約/Pubmed全文|CrossRef全文
76. Wu G,Ai T,Kim JJ,Mohapatra B,Xi Y,Li Z,et al. アルファ1syntrophinの突然変異および長いQTシンドローム:ナトリウムチャネルの中断の病気。 (2008)1:193-201. 土井:10.1161/CIRCEP.108.769224
Pubmed要約/Pubmed全文|CrossRef全文
77. Yang Y,Yang Y,Liang B,Liu J,Li J,Grunnet M,et al. Kir3の識別。先天性QT延長症候群における4つの突然変異。 A M J Hum Genet(2 0 1 0)8 6:8 7 2−8 0. 土井:10.1016/j.ajhg.2010.04.017
Pubmed要約/Pubmed全文|CrossRef全文
78. ローデンDM. 不整脈のメカニズムと管理。 Am J Cardiol(1998)82:49I–57i.doi:10.1016/S0002-9149(98)00472-X
CrossRef全文
79. Mitcheson JS,Chen J,Lin M,Culberson C,Sanguinetti MC. 薬物誘発性QT延長症候群の構造的基礎。 Proc Natl Acad Sci U S A(2 0 0 0)9 7:1 2 3 2 9−3 3. ドイ:10.1073/pnas.210244497
クロスリファレンス全文
80. Kanankeril PJ,Roden DM. 薬物誘発性qt延長およびtorsade de pointes:最近の進歩。 Curr Opin Cardiol(2007)22:39-43. doi:10.1097/HCO。0b013e32801129eb
Pubmed要約/Pubmed全文|CrossRef全文
81. Veerman CC,Verkerk AO,Blom MT,Klemens CA,Langendijk PN,van Ginneken AC,et al. 遅い遅延整流器カリウム電流遮断は、薬物誘発性QT延長症候群に重要な貢献しています。 2013年6月10日から9日まで放送された。 土井:10.1161/CIRCEP.113.000239
Pubmed要約/Pubmed全文|CrossRef全文
82. Sesti F,Abbott GW,Wei J,Murray KT,Saksena S,Schwartz PJ,et al. 抗生物質誘発性心臓不整脈に関連する一般的な多型。 Proc Natl Acad Sci U S A(2 0 0 0)9 7:1 0 6 1 3−8. ドイ:10.1073/pnas.180223197
Pubmed要約/Pubmed全文|CrossRef全文
83. Kääb S,Crawford DC,Sinner MF,Behr ER,Kannankeril PJ,Wilde AA,et al. 大規模な候補遺伝子の調査は、薬物誘発torsadesドpointesの可能なモジュレーターとしてKCNE1D85N多型を識別します。 2012年5月9日から9月9日まで放送された。 土井:10.1161/CIRCGENETICS.111.960930
Pubmed要約/Pubmed全文|CrossRef全文
84. 中村K,片山Y,草野KF,原岡K,谷Y,長瀬S,et al. 抗KCNH2抗体誘発性QT延長症候群:qt延長症候群の新規な後天性形態。 J Am Coll Cardiol(2007)50:1808-9. 土井:10.1016/j.jacc.2007.07.037
クロスリファレンス全文
85. Moss AJ,Zareba W,Benhorin J,Locati EH,Hall WJ,Robinson JL,et al. 遺伝性qt延長症候群の遺伝的に異なる形態のECG T波パターン。 Circulation(1 9 9 5)9 2:2 9 2 9−3 4. 土井:10.1161/01.CIR92.10.2929
Pubmed要約|Pubmed全文|CrossRef全文
86. Zhang L,Timothy KW,Vincent GM,Lehmann MH,Fox J,Giuli LC,et al. 先天性QT延長症候群におけるST-T波パターンと再分極パラメータのスペクトル:ECG所見は遺伝子型を同定する。 循環(2000)102:2849-55。 土井:10.1161/01.CIR102.23.2849
Pubmed要約/Pubmed全文|CrossRef全文
87. Tan HL,Bardai A,Shimizu W,Moss AJ,Schulze-Bahr E,Noda T,et al. 先天性QT延長症候群における不整脈の遺伝子型特異的発症:治療の可能性への影響。 循環(2006)114:2096-103. 土井:10.1161/106.642694
Pubmed要約/Pubmed全文|CrossRef全文
88. ロカティEH,Zareba W,Moss AJ,Schwartz PJ,Vincent GM,Lehmann MH,et al. 先天性qt延長症候群の患者における臨床症状における年齢および性別関連の違い:国際LQTSレジストリからの所見。 Circulation(1 9 9 8)9 7:2 2 3 7−4 4. 土井:10.1161/01.CIR97.22.2237
Pubmed要約|Pubmed全文|CrossRef全文
89. Zareba W,Moss AJ,Schwartz PJ,Vincent GM,Robinson JL,Priori SG,et al. Qt延長症候群の臨床経過に及ぼす遺伝子型の影響。 国際QT延長症候群レジストリ研究グループ。 N Engl J Med(1 9 9 8)3 3 9:9 6 0−5. 土井:10.1056/NEJM199810013391404
Pubmed要約/Pubmed全文|CrossRef全文
90. Goldenberg I,Moss AJ,Bradley J,Polonsky S,Peterson DR,McNitt S,et al. 40歳以降のQT延長症候群。 循環(2008)117:2192-201。 土井:10.1161/107.729368
Pubmed要約/Pubmed全文|CrossRef全文
91. Zareba W,Moss AJ,Locati EH,Lehmann MH,Peterson DR,Hall WJ,et al. シンドロームの登録簿。 遺伝子型によるQT延長症候群の臨床経過に及ぼす年齢および性別の影響を調節する。 J Am Coll Cardiol(2003)42:103-9. ドイ:10.1016/S0735-1097(03)00554-0
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
92. Seth R,Moss AJ,McNitt S,Zareba W,Andrews ML,Qi M,et al. QT延長症候群および妊娠。 J Am Coll Cardiol(2007)49:1092-8. 土井:10.1016/j.jacc.2006.09.054
Pubmed要約/Pubmed全文|CrossRef全文
93. Schwartz PJ,Priori SG,Dumaine R,Napolitano C,Antzelevitch C,Stramba-Badiale M,et al. 乳幼児突然死症候群とQT延長症候群との間の分子リンク。 N Engl J Med(2 0 0 0)3 4 3:2 6 2−7. doi:10.1056/NEJM200007273430405
CrossRef全文
94. Schwartz PJ,Priori SG,Bloise R,Napolitano C,Ronchetti E,Piccinini A,et al. 乳幼児突然死症候群の小児における分子診断。 Lancet(2 0 0 1)3 5 8:1 3 4 2−3. ドイ:10.1016/S0140-6736(01)06450-9
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
95. Christiansen M,Tønder N,Larsen LA,Andersen PS,Simonsen H,Oyen N,et al. HERG K+イオンチャネルにおける変異:QT延長症候群と乳児突然死症候群との間の新規なリンク。 A M J Cardiol(2 0 0 5)9 5:4 3 3−4. 土井:10.1016/j.amjcard.2004.09.054
Pubmed要約/Pubmed全文|CrossRef全文
96. Djはアッカーマン-MJ。 若いの突然の原因不明の死のための死後のQT延長症候群の遺伝子検査。 J Am Coll Cardiol(2007)49:240-6. 土井:10.1016/j.jacc.2006.10.010
Pubmed要約/Pubmed全文|CrossRef全文
97. Hofman N,Tan HL,Clur SA,Alders M,van Langen IM,Wilde AA. 小児期における突然の心臓死に対する遺伝性心臓病の寄与。 Pediatrics(2007)120:e967–73. ドイ:10.1542/peds.2006-3751
Pubmed要約/Pubmed全文|CrossRef全文
98. Wedekind H,Smits JP,Schulze-Bahr E,Arnold R,Veldkamp MW,Bajanowski T,et al. 乳児突然死の早期発症に関連するSCN5A遺伝子におけるDe novo変異。 循環(2001)104:1158-64. doi:10.1161/hc3501.095361
CrossRef全文
99. Wang DW,Desai RR,Crotti L,Arnestad M,Insolia R,Pedrazzini M,et al. 乳幼児突然死症候群における心臓ナトリウムチャネル機能不全。 循環(2007)115:368-76. 土井:10.1161/106.646513
Pubmed要約/Pubmed全文|CrossRef全文
100. Van Norstrand DW,Valdivia CR,Tester DJ,Ueda K,London B,Makielski JC,et al. 乳幼児突然死症候群における新規グリセロール-3-リン酸デヒドロゲナーゼ1様遺伝子(GPD1-L)変異の分子的および機能的特性評価。 循環(2007)116:2253-9. 土井:10.1161/107.704627
Pubmed要約/Pubmed全文|CrossRef全文
101. Tan BH,Pundi KN,Van Norstrand DW,Valdivia CR,Tester DJ,Medeiros-Domingo A,et al. ナトリウムチャネルベータサブユニットにおける乳幼児突然死症候群関連変異。 心のリズム(2010)7:771-8. 土井:10.1016/j.hrthm.2010.01.032
Pubmed要約/Pubmed全文|CrossRef全文
102. Miller TE,Estrella E,Myerburg RJ,Garcia de Viera J,Moreno N,Rusconi P,et al. QT延長シンドロームのための再発第三期の胎児の損失そして母性的なmosaicism。 循環(2004)109:3029-34. 土井:10.1161/01.CIR0000130666.81539.9E
Pubmed要約/Pubmed全文|CrossRef全文
103. Chockalingam P,Crotti L,Girardengo G,Johnson JN,Harris KM,van der Heijden JF,et al. すべてのベータ遮断薬がqt延長症候群のタイプ1および2の管理において等しいわけではありません:メトプロロールの下での事象のより高い再発。 J Am Coll Cardiol(2012)60:2092-6. 土井:10.1016/j.jacc.2012.07.046
Pubmed要約/Pubmed全文|CrossRef全文
104. Schwartz PJ,Priori SG,Cerrone M,Spazzolini C,Odero A,Napolitano C,et al. QT延長症候群の影響を受けた高リスク患者の管理における左心臓交感神経脱神経。 循環(2004)109:1826-33. 土井:10.1161/01.CIR0000125523.14403.1E
Pubmed要約/Pubmed全文|CrossRef全文
105. Singh B、al Shahwan SA、Habbab MA、al Deeb SM、Biary N.特発性QT延長症候群:正しい質問をする。 ランセット(1993)341:741. 土井:10.1016/0140-6736(93)90501-7
CrossRef全文
106. Gorgels AP、Al Fadley F、Zaman L、Kantoch MJ、Al Halees Z.房室伝導障害を伴うQT延長症候群:乳児の悪性変異体。 J Cardiovasc Electrophysiol(1 9 9 8)9:1 2 2 5−3 2. ドイ:10.1111/j.1540-8167.1998.00096x
Pubmed要約/Pubmed全文|CrossRef全文
107. Kantoch MJ、Kurashi MM、Bulbul ZR、Gorgels AP。 複雑な先天性心疾患、房室ブロック、およびtorsade de pointes心室頻脈を有する新生児。 Pacing Clin Electrophysiol(1 9 9 8)2 1:2 6 6 4−7. ドイ:10.1111/j.1540-8159.1998.tb00043.x
クロスリファレンス全文
108. エル-Hazmi MA、アル-Swailem AR、Warsy AS、アル-Swailem AM、Sulaimani R、アル-Meshari AA。 サウジアラビアの人口の中で同族。 J Med Genet(1 9 9 5)3 2:6 2 3−6. ドイ:10.1136/jmg.32.8.623
クロスリファレンス全文
109. El Mouzan MI,Al Salloum AA,Al Herbish AS,Qurachi MM,Al Omar AA. サウジの子どもたちの血縁と主要な遺伝性疾患:コミュニティベースの断面研究。 Ann Saudi Med(2008)28:169-73. 土井:10.4103/0256-4947.51726
Pubmed Abstract|Pubmed Full Text|CrossRef Full Text
110. Medeiros-Domingo A,Bhuiyan ZA,Tester DJ,Hofman N,Bikker H,van Tintelen JP,et al. カテコールアミン作動性多型心室頻拍または遺伝子型陰性、運動誘発性QT延長症候群のいずれかで以前に診断された患者におけるryr2エンコードされた: 広範囲の開いた読書フレームの突然変異の分析。 J Am Coll Cardiol(2009)54:2065-74. 土井:10.1016/j.jacc.2009.08.022
Pubmed要約/Pubmed全文|CrossRef全文
111. Al-Aama JY,Al-Ghamdi S,Bdier AY,Wilde AA,Bhuiyan ZA. JervellおよびLange-Nielsen症候群の原因となるKCNQ1遺伝子のDe novo変異。 2013年、クリン-ジュネット(Clin Genet)に移籍。 doi:10.1111/cge。12300
Pubmed要約/Pubmed全文|CrossRef全文
112. Splawski I,Timothy KW,Tateyama M,Clancy CE,Malhotra A,Beggs AH,et al. SCN5Aナトリウムチャネルのバリアントは、心臓不整脈のリスクに関与しています。 Science(2002)297:1333-6. 土井:10.1126/科学。1073569
クロスリファレンス全文
113. ワイルドAA、ベッツィーナCR。 心臓不整脈の遺伝学。 Heart(2 0 0 5)9 1:1 3 5 2−8.