Article
Josivan Gomes Lima1*,Marcel Catão Ferreira dos Santos1,Julliane Tamara Arañjo de Melo Campos2
1departamento de medicina clínica,disciplina de endocrinologia e metabologia. 病院Universitário Onofre Lopes,Universidade Federal do Rio Grande do Norte(UFRN),Natal,Rn,Brazil
2Trairi保健科学学部,Federal University of Rio Grande do North(UFRN),Natal,Rn,Brazil
Abstract
先天性全身性脂肪異栄養症(CGL)はまれで重度の常染色体劣性疾患である….. 患者は体脂肪の貯蔵に欠陥があり、その結果、異所性組織、主に肝臓に脂肪を沈着させ、肝硬変を発症する可能性がある。 インスリン抵抗性は典型的な発見であり、インスリンの高い毎日の用量を必要とする糖尿病を引き起こす。 ブラジルのリオグランデ-ド-ノルテ州では、CGL患者の最大のコホートの一つを持っています。 本稿では,この疾患の病態生理,臨床像および治療についてレビューした。
はじめに
2型糖尿病は世界的な健康上の問題であり、通常、過剰な体重と内臓脂肪の増加により末梢インスリン抵抗性が生じ、膵臓がインスリンを放出してこの抵抗性を補うことができないことが原因である。 他のあまり一般的でないタイプの糖尿病は、Berardinelli-Seip先天性脂肪異栄養症(BSCL)としても知られている先天性全身性脂肪異栄養症(CGL)のような特定の遺伝的変異 CGLは,遺伝子変異に基づいて四つのタイプに分類される常染色体劣性疾患である。 変えられた遺伝子はadipocyteの中のadipocyteの形成、脂質の生産および適切な貯蔵のための必要な機能を担います。 変異は脂肪組織を減少させ、脂肪肝、炭水化物代謝の変化、高インスリン血症および先端メガロイド特徴を伴う重度のインスリン抵抗性、および脂質異常症1-3を引き起こす。 CGL症候群は、世界で報告されている約500例を持っています。 ブラジルでは、リオグランデ-ド-ノルテ州(RN)で、過去20年間に54例の診断、治療、フォローを行っています4、5。 二次データを使用して記述的な研究では、我々は103RN6の患者の合計を推定しました。 これは、文献(で報告されたものよりもはるかに高い有病率を示しています1: 1万円)7.
脂質液滴におけるトリアシルグリセロールの形成と貯蔵
トリグリセリドとリン脂質の生合成(図1A)は、グリセロール-3-リン酸アシルトランスフェラーゼ(GPAT)が1位のグリセロール-3-リン酸アシルトランスフェラーゼをアシル化し、1-アシルグリセロール-3-リン酸(リゾホスファチジン酸)を形成することから始まる。 反応式の通り、1,2-ジアシルグリセロール-3-リン酸アシルトランスフェラーゼ(1-Acylglycerol-3-phosphate acyltransferase、1,2-Diacylglycerol-3-phosphate acyltransferase)は、次の化学反応を触媒する酸化還元酵素である。 それはトリグリセリドおよびphosphoglycerides両方の生合成の細道の主中間ステップです。 AGPAT酵素には11種類のアイソフォームがあり、異なる遺伝子によってコードされている4。 AGPAT1およびAGPAT2は最も広く調査されます。 AGPAT1は、精巣、膵臓、および脂肪組織および心臓、胎盤、脳、肺のような他の組織に高レベルで存在するが、AGPAT2は脂肪組織に豊富である。 以下のステップでは、細胞質酵素ホスファチジン酸ホスファターゼ(PAPまたはlipin)は1,2-ジアシルグリセロールを起源とし、1,2-ジアシルグリセロールアシルトランスフェラーゼ(DGAT)はトリアシルグリセロール4を形成する。 ホスファチジン酸およびジアシルグリセロールはまた、カルジオリピン、ホスファチジルイノシトール、ホスファチジルコリンなどの他のリン脂質を起源とすることができる。
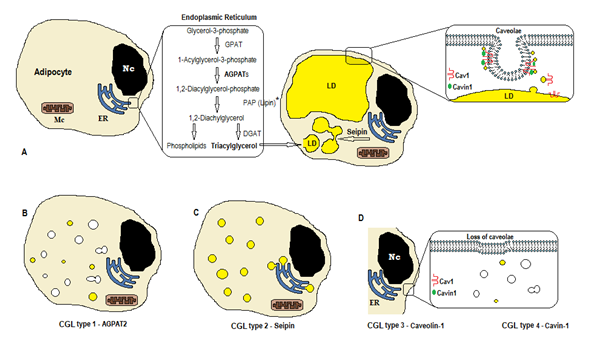
図1. Cglタイプによるトリグリセリド合成のスキーム。 (A)脂肪細胞におけるトリアシルグリセロール(TAG)の正常な合成および貯蔵。 (B)AGPAT2の突然変異は、タグ産生を減少させる(一部は、他のAgpatの刺激下でまだ合成される)。 (C)seipin遺伝子の変異はタグ合成および脂質滴(L d)形成および融合を減少させる。 (D)カベオリン−1およびカビン−1は、カベオラエの形成および安定化のために必要である。 CAV1(タイプ3)またはCAVIN1(タイプ4)の突然変異により膜でcaveolaeの損失を引き起こすことができます。 Nc、核。 小胞体小胞体小胞体小胞体小胞体小胞体小胞体 Mc、ミトコンドリア… *LipinはERのseipinによって固定される細胞質の酵素です。
これらの反応は、脂肪細胞の小胞体(ER)で起こり、トリグリセリドの進行性蓄積が小さな脂質小滴(LD)の形成を引き起こす8。 遺伝子BSCL2の産物は、大きなLDを起源とする小さなLDの融合を引き起こすseipinと呼ばれる膜貫通タンパク質である。 SeipinはERに存在し、ERとLDとLD9のトリグリセリドの結合間の脂質の交通を促進する新生LDとの接続点に集中します。 Seipinはまた細胞質酵素lipin1へERのアンカーとして機能するかもしれません。 セイピンは、脂質液滴の融合、サイズ、および形態に必要であることに加えて、脂肪形成(リピン1との相互作用を介して)および細胞トリグリセリドlipolysis10,11にも不可欠である10,11。 セイピンの欠乏は、前脂肪細胞の脂肪細胞への分化を妨げ、最終的な成熟に影響を与える9、BSCL2ノックアウトを有する間葉系幹細胞の研究によって示されているように12。 非脂肪組織はまた、セイピンを発現し、他の機能が決定されるべきである。
脂肪細胞では、50-100nmの膜陥入に特化したカベオラエが原形質膜面積の20%を占め、脂肪細胞をカベオラエの最も密度の高い細胞としている13。 脂質小滴の形成には、膜タンパク質(カベオリン-カベオラエ膜の主成分)および細胞質タンパク質(Cavin-1)が必要である14。 遺伝子CAV1、CAV2、およびCAV3は、同様の構造(それぞれCaveolin-1、Caveolin-2、およびCaveolin-3)を有するカベオリンの三つの形態をコードする。 カベオリン-1およびカベオリン-2は、脂肪細胞、線維芽細胞、および内皮細胞に存在し、カベオリン-3は骨格筋および心臓筋にのみ存在する13,15。 Caveolin-1は最も重要で最も研究されています。 それは2つの異なるアイソフォーム(1aと1b)で表されます。 カベオリン-1は原形質膜から脂質液滴に移動し、脂質輸送および代謝に必要である16。 脂質のしぶきは供給の後でトリグリセリドを貯え、これらの分子は脂肪酸に加水分解され、絶食の間に解放されます;このメカニズムはCaveolin-116によって調 カベオリン-1欠乏症はまた、オートファジーによる細胞死に対する感受性を増加させる17。
遺伝子CAVIN1は、caveolae associated protein1(Cavin-1)14,16と呼ばれる細胞質タンパク質をコードしており、これはcaveolaeの形成と安定化に必須である。 Cavin−1は、脂肪細胞、筋肉細胞、および他の細胞で発現され、caveolae由来のシグナルの伝達にも不可欠である1 4、1 8。 CAVIN1のノックアウトはmuscle14を含むすべてのティッシュでcaveolaeの不在を引き起こす一方、CAV1遺伝子のノックアウトは非筋肉細胞でcaveolaeの欠乏を引き起こ Caveolaeの欠乏は他の細道の脂肪分解、脂肪酸の変化、トリグリセリドの統合および信号の規則に影響を与えることができます。
CGLの種類
検出可能な遺伝的変化に基づいて、四つのタイプが記載されています。 タイプ1および2はケースの95%に責任があり、タイプ2により深刻な影響を受けた表現型があります。 タイプ3の一つのケースと約30のタイプ4のケースが報告されている4。
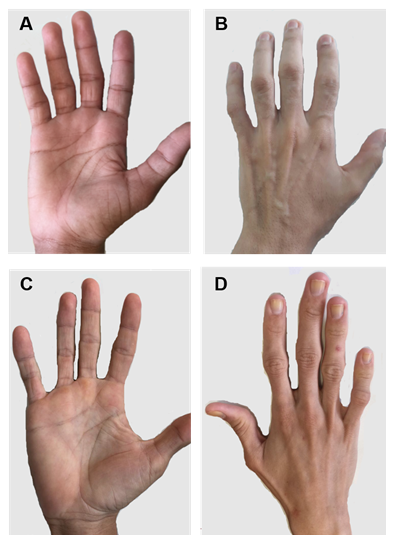
図2. CGLタイプ1および2の患者の手。 (A)および(B)タイプ1患者の手の前方および後方の眺め。 明らかに正常な手、まだ機械的な脂肪組織があるので。 (C)および(D)タイプ2患者の手の前方および後方の眺め。 この疾患の重症度はより高く、脂肪の欠如は明らかであり、容易に顕著である。
CGLタイプ1。 1999年、Garg et al. 染色体9q34上の患者の変異を記載し、三年後Agarwal et al. この変異の影響を受けた酵素としてAGPAT2を示した2,19。 このAGPAT2の突然変異が原因で、トリアシルグリセロールのどれもまたは最低の生産は他のアイソフォームの刺激によって起こりません。 AGPAT2ノックアウトマウスの表現型は、cgl型のヒトの表現型と同様であり、病態生理におけるこの酵素の役割を確認している20,21。
CGLタイプ2。 マグレ他 seipin遺伝子(染色体11q13)の変異を最初に同定した3人であった。 セイピン遺伝子(BSCL2)の変異(主にナンセンス)は、切り捨てられたタンパク質を生成し、異なるメカニズムによって脂質代謝に影響を与えることができます:a)セイピン安定性の低下、b)リピン1を結合する能力の低下、およびc)オリゴマー化し、ER膜に排他的にそれ自体を局在化する失敗11。 いくつかの細胞はまだトリアシルグリセロールおよび小さな脂質小滴を生成することができるが、大きな脂質小滴は、これらの小さな脂質小滴の融合 また、ペルオキシソーム増殖剤活性化受容体ガンマ(PPARG)、ならびにアディポネクチンおよび脂肪細胞脂肪酸結合タンパク質(FABP4)などの脂肪形成因子の発現にも障害がある11、16。 Seipinの不足はadipogenesisを損ない、脂肪分解を高め、そしてadipocytesのトリグリセリドの蓄積を防ぎます。
CGLタイプ3。 このタイプは、CGL表現型を有するにもかかわらず、遺伝子AGPAT2またはBSCL222に変異を有さなかった患者において最近記載された。 Cav1の突然変異のマウスは食事療法誘発の肥満に対して抵抗力があり、インシュリン抵抗性、hypertriglyceridemia、減らされたadiponectin、減らされた脂肪質の固まりおよび小さいadipocytes16があ マウスでの研究に基づいて候補遺伝子を選択した後、Kim et al. 染色体7q3122上のカベオリン-1遺伝子(CAV1)におけるナンセンス変異の存在を確認した。
CGLタイプ4。 これはまれなタイプであり、影響を受けた遺伝子はタンパク質Cavin-1をコードするCAVIN1である。 ヒトでは、一般化された先天性脂肪異栄養症および筋ジストロフィーの患者で報告されている15,23。
最近、PCYT1AおよびPPARG遺伝子の変異がリポジストロフィーを引き起こすことも報告されている24,25。
臨床的特徴
CGL患者は、通常、先端巨大症、黒皮症、phebomegaly、肝腫大、および筋肉肥大を示す5,26,27。 いくつかの著者は、臍ヘルニアを症候群の臨床所見として引用している26。 私たちは一連の患者でそれの頻度を評価しました、そして、それらのどれもこの変化を提示しませんでした28。 実際には、臍周囲脂肪組織の欠如は、臍瘢痕の突出を引き起こし、これは誤ってヘルニアと診断される可能性があります28,29。
脂肪細胞が十分に脂肪を貯蔵できなくなると、肝臓や筋肉などの他の組織に蓄積し、重度のインスリン抵抗性を引き起こす。 骨密度測定(DXA)は、正常または高い骨鉱物密度を示すことがあります30および総体脂肪の減少(通常6%未満)27。 低体脂肪の結果として、血清アディポネクチンとレプチンも低い27。 レプチンは飢餓を制御するのに不可欠であるため、これらの患者は典型的には過食症を有し、これは小児期から容易に明らかである。 アディポネクチンはインスリン感作剤として重要な役割を果たし,その欠乏はインスリン抵抗性を悪化させる。 それにもかかわらず、最初は、グルコースおよび糖化ヘモグロビンは、非常に高いインスリンレベルを犠牲にして正常である。 糖尿病は通常思春期に始まり、私たちのシリーズでは、発症の平均年齢は15.8±7.1歳であった27。 当初、それらは経口薬で制御され、数年後に高用量のインスリンを必要とする27。 動脈性高血圧症は、患者の三分の一に起こる27。
各CGLタイプにはいくつかの特定の臨床的特徴があります。 1型の患者は、特に手のひら、靴底、眼窩、関節周囲領域に依然として機械的脂肪を示す31。 対照的に、2型患者は代謝および機械的脂肪組織の欠如を示す。 セイピンは脳や小脳で高度に発現し、神経機能の調節にも関与しています。 2型患者の半数以上が何らかの認知障害を持っている1,8。 タイプ3および4は機械的および骨髄脂肪の保存を有し、タイプ4は高い血清クレアチンキナーゼおよび脊髄不安定性に関連する筋力低下を有する15。
性別特異的な臨床的特徴もある。 多嚢胞性卵巣および無月経が一般的である32。 月経周期は通常、おそらくインスリン感受性の改善およびLH脈動の回復のために、メトレプチンの使用により正常に戻る32。 2型の男性は、生殖細胞にセイピンがないために奇形性精子症を有することができる33。
高トリグリセリド血症は人生の最初の年から発生し、急性膵炎を引き起こす可能性があります。 HDLは通常30mg/dLより低いです。 肝臓酵素の上昇も初期の発見であり、肝臓の脂肪沈着に由来する。 血清血小板の漸進的な減少は、肝疾患および肝硬変の可能性のある悪化を示唆する34。
Cavin-1は筋肉細胞に存在するため、4型の患者は軽度の筋力低下とクレアチンキナーゼの上昇15を有する。
2型を中心とした平均余命は大幅に減少し、30歳までに死亡することはまれではない(過去20年間に死亡した19人の患者に基づく個人的な経験)。 死因は、糖尿病(腎不全、突然死)、肝臓(肝硬変、消化出血)または感染症に関連しています。
診断と治療
CGL診断は臨床データに基づいています:先端巨大症の特徴、黒皮症、総体脂肪の減少、筋肉肥大、および臍帯瘢痕の突出。 また、実験室のデータは、重度のインスリン抵抗性および高トリグリセリド血症を伴う糖尿病を示すことができる。 画像検査は、主に肝臓および膵臓における脂肪の異所性沈着物(肝腫大および膵臓脂肪症を伴う肝脂肪症)を同定するのに役立ち得る。 DXAは低い体脂肪および高い骨density30を確認できます。
CGLの治療は、脂肪、主にトリグリセリドおよび併存疾患を予防および制御するための血糖指数の高い食品の摂取量を減少させた食事の厳格な管理29。 但し、理想的な食事療法は高められた食欲および支持される厳しい制限のために達成する挑戦的な目的である。 重度の心筋症などの禁忌を有する患者を除いて、併存疾患の制御を改善するために身体活動も奨励されるべきである29。
薬物治療に関しては、これらの患者は、糖尿病、高血圧、および脂質異常症のガイドラインのための通常の薬物療法で治療することができます。 糖尿病およびインスリン抵抗性の治療のための最初の選択はメトホルミンであるが、通常、それは十分ではない。 部分的な脂肪異栄養症の治療とは異なり、チアゾリジンジオンは注意を払って使用すべきである29。 他の経口抗糖尿病薬が使用されるが、それらはCGL患者において特異的に研究されなかった。 SGLT2阻害剤(ダパグリフロジン)の使用は、心筋症を予防する利点を有することができることを示唆している動物のデータがあります35;研究は、ヒトでこ 病気が進歩し、厳しいインシュリン抵抗性が起こると同時に、インシュリンの高い毎日の線量は必要です。 皮下脂肪組織の欠如は、高用量のインスリンを投与する際の問題である。 より濃縮されたインスリン(U-300またはU500)が必要な場合がある36。 これらの患者は、主にトリグリセリドの増加および低HDLのために重度の脂質異常症を呈し、したがって、急性膵炎を予防するためにフィブラートの使用 さらに、これらの患者の高い心血管リスクのために、スタチンによる介入を考慮すべきであり、LDLまたは非HDLの標的は厳密でなければならない29。
メトレプチンの毎日の注入は食欲の重要な減少を引き起こし、glycemia、トリグリセリド血症およびレバー酵素の低下によって利点を持って来ます。 特に小児では、おそらく肝腫大の減少による腹囲の減少が注目に値する。
結論
CGLは、糖尿病(通常は高用量のインスリンを必要とする)および早期死亡で起こり得るまれで重度の疾患である。 患者の表現型は非常に特徴的であり、しかし、早期診断を行うために医療専門家による症候群の知識を必要とする。 Metreleptinは病気の自然史を変更できる唯一の薬物現時点でのようです。
利益相反:なし。
- Nolis T.より一般的な遺伝的および後天性脂肪質の背後にある病態生理を探索する。 人間の遺伝学のジャーナル。 2014Jan;59(1):16-23.
- Agarwal AK,Arioglu E,De Almeida S,et al. AGPAT2は染色体9q34にリンクされている先天性全身性脂肪異栄養症で変異しています。 ナットジェネット 2002May;31(1):21-3.
- Magre J,Delepine M,Khallouf E,et al. 染色体11q13上のBerardinelli-Seip先天性脂肪異栄養症で変化した遺伝子の同定。 自然の遺伝学。 2001Aug;28(4):365-70.
- Patni N,Garg A. 先天性全身性脂肪変性症-代謝機能障害への新しい洞察。 自然のレビュー内分泌学。 2015Sep;11(9):522-34.
- ガーグA.はリポジストロフィーを獲得し、継承した。 医学のニューイングランドジャーナル。 2004Mar18;350(12):1220-34.
- de Azevedo Medeiros LB,Candido Dantas VK,Craveiro Sarmento AS,et al. ブラジル北東部のリオグランデ-ド-ノルテ州におけるBerardinelli-Seip先天性脂肪異栄養症の有病率が高い。 ダイアベトールメタボシンド… 2017; 9: 80.
- Chiquette E,Oral EA,Garg A,et al. 一般化された部分的な脂肪異栄養症の有病率の推定:所見と課題。 糖尿病、メタボリックシンドロームおよび肥満:ターゲットおよび療法。 2017: 375-83.
- Wee K,Yang W,Sugii S,et al. 脂肪異栄養症およびseipin機能の機械論的理解の方。 バイオサイエンスレポート。 2014; 34(5).
- Dollet L,Magre J,Cariou B,et al. Seipinの機能:Bscl2/seipinのノックアウトのマウスモデルからの新しい洞察力。 ビオチミー 2014Jan;96:166-72.
- Sim MF,Dennis RJ,Aubry EM,et al. 人間のlipodystrophy蛋白質seipinはadipogenic PAのホスファターゼのlipin1のためのERの膜のアダプターです。 分子代謝。 2012; 2(1): 38-46.
- Sim MF,Talukder MM,Dennis RJ,et al. ヒト脂肪異栄養タンパク質seipinにおける自然発生する変異の分析は、複数の潜在的な病原性メカニズムを明らかにする。 “糖尿病” 2013Nov;56(11):2498-506.
- Payne VA,Grimsey N,Tuthill A,et al. ヒト脂肪異栄養遺伝子BSCL2/seipinは、正常な脂肪細胞分化に不可欠である可能性があります。 糖尿病 2008Aug;57(8):2055-60.
- Cohen AW,Hnasko R,Schubert W,et al. 健康および病気におけるカベオラエおよびカベオリンの役割。 生理的なレビュー。 2004Oct;84(4):1341-79.
- Pilch PF,Liu L.Fat caves:caveolae,脂質輸送および脂肪細胞における脂質代謝。 内分泌学および代謝の動向:TEM。 2011Aug;22(8):318-24.
- 林YK,松田C,小川M,et al. ヒトPTRF変異は、一般化された脂肪異栄養症を伴う筋ジストロフィーをもたらすカベオリンの二次欠損を引き起こす。 J-クリニーク-インベストメント。 2009Sep;119(9):2623-33.
- パートンRG、デルポゾMA。 細胞膜センサー、保護装置およびオルガナイザーとしてCaveolae。 自然は分子細胞生物学をレビューします。 2013Feb;14(2):98-112.
- Le Lay S,Briand N,Blouin CM,et al. 脂肪栄養caveolin-1欠損マウスモデル成熟した脂肪細胞のオートファジーを明らかにします。 オートファジー 2010Aug;6(6):754-63.
- Liu L,Brown D,McKee M,et al. Cavin/PTRFの欠失によりcaveolae、dyslipidemiaおよびブドウ糖の不耐性の全体的な損失を引き起こします。 細胞の代謝。 2008年8月(4):310-7。
- Garg A,Wilson R,Barnes R,et al. 先天性全身性脂肪異栄養症の遺伝子は、ヒト染色体9q34にマップされます。 臨床内分泌および代謝のジャーナル。 1 9 9 9年9月;8 4(9):3 3 9 0−4.
- Vogel P,Read R,Hansen G,et al. Agpat2-/-マウスにおける先天性全身性脂肪異栄養症の病理。 獣医病理学。 2011May;48(3):642-54.
- Cortes VA,Curtis DE,Sukumaran S,et al. 先天性全身性脂肪異栄養症のAGPAT2欠損マウスモデルにおける肝脂肪症とインスリン抵抗性の分子機構。 細胞の代謝。 2009Feb;9(2):165-76.
- Kim CA,Delepine M,Boutet E,et al. Berardinelli-Seip先天性脂肪異栄養症とホモ接合ナンセンスcaveolin-1変異の関連付け。 Jクリノールメタブ… 2008Apr;93(4):1129-34.
- Rajab A,Straub V,McCann LJ,et al. PTRF-CAVIN変異による筋波紋(CGL4)を伴う先天性全身性脂肪異栄養症の新しい形態における致命的な心臓不整脈およびQT延長症候群。 PLoSの遺伝学。 2010Mar12;6(3):e1000874.
- Payne F,Lim K,Girousse A,et al. 先天性脂肪異栄養症および脂肪肝疾患を有するヒトにおけるケネディホスファチジルコリン経路を破壊する変異。 Proc Natl Acad Sci U S A.2014Jun17;111(24):8901-6.
- Dyment DA,Gibson WT,Huang L,et al. PPARGの二対立遺伝子変異は,Berardinelli-Seip症候群と同様の先天性の全身性脂肪異栄養症を引き起こす。 ユーロJ Med Genet. 2014Sep;57(9):524-6.
- Garg A.Clinical review#:Lipodystrophies:遺伝的および後天的な体脂肪障害。 臨床内分泌および代謝のジャーナル。 2011Nov;96(11):3313-25.
- Lima JG,Nobrega LH,de Lima NN,et al. 先天性全身性脂肪異栄養症を有する大規模な一連の患者の臨床および検査データ。 ダイアベトールメタボシンド… 2016; 8: 23.
- Lima GJ,Lima NN,Oliveira CF,et al. Berardinelliseip症候群の患者の臍ヘルニア:それは本当にヘルニアですか。 Jクリン-モレノールエドクリノールエドクリノールエ 2015; 1(1): 3.
- ブラウンRJ,Araujo-Vilar D,Cheung PT,et al. 脂肪異栄養症候群の診断と管理:マルチ社会実践ガイドライン。 Jクリノールメタブ… 2016Dec;101(12):4500-11.
- Lima JG,Nobrega LH,Lima NN,et al. Berardinelli-Seip先天性脂肪異栄養症患者の骨密度は、小柱部位および2型患者でより高い。 J-クリント-デンシトム 2016年11月25日に発売された。
- Simha V,Garg A.AGPAT2またはseipin遺伝子の変異によって引き起こされる先天性全身性脂肪異栄養症患者における体脂肪分布における表現型の異質性。 Jクリノールメタブ… 2003年11月;88(11):5433-7。
- Musso C,Cochran E,Javor E,et al. 男性および女性のhypoleptinemic lipodystrophic患者の女性および下垂体機能のhyperandrogenismそしてmenstrual機能に対する組換えのメチオニルの人間のレプチン療法の長期効果。 メタボリックシンドロームとは? 2005Feb;54(2):255-63.
- Jiang M,Gao M,Wu C,et al. 精巣セイピンの欠如は、男性の奇形精子症症候群を引き起こす。 2014年5月13日;111(19):7054-9.
- Mitchell O,Feldman DM,Diakow M,et al. 慢性肝疾患における血小板減少症の病態生理。 ヘパット-メッド 2016; 8: 39-50.
- Joubert M,Jagu B,Montaigne D,et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes. 2017 Apr; 66(4): 1030-40.
- Lima JG, Lima NN, Lima RLM, et al. Glargine U300 Insulin as a Better Option than Degludec U100 to Treat a Congenital Generalized Lipodystrophy Patient. Clin Diabetes Res. 2017; 1(1).