Frontiers in Neurology
Introduction
Neuroacanthocytosisは、棘状変形赤血球(アカントサイト)と組み合 このレポートは、Vps13A(空胞タンパク質ソート13ホモログA)染色体9q(1)上の遺伝子の常染色体劣性変異によって引き起こされる世界中の推定1,000影響を受けた個人と孤児疾患である舞踏病-アカントサイトーシス(ChAc)に焦点を当てています。 疾患は進行性のコースを実行し、増加した死亡率の原因は、嚥下障害などのリスク条件と相関運動機能を低下させることができるだけでなく、突然の不 これまでのところ、原因となる治療法はありません。
アカントサイトーシスと運動障害の疑わしい共存は、1970年代にLevineとCritchley(4、5)によって最初に報告され、以来、この疾患の顕著な特徴として認められてきた。 しかし、我々のケースレポートでは、臨床プレゼンテーションの広い多様性を示唆し、明らかな運動障害を欠いているChAcのまれな臨床表現型を記述します。 診断ツールは、正しい診断を確立するためにこれらの課題を満たす必要があり、我々はここで重要な方法として分子神経イメージングを提案します。
症例報告
男性指数患者は、25歳で最初に二つの言われていない両側強直間代発作を提示しました。 病歴はなかった。 その年齢の脳MRIスキャンおよびEEGは正常であり、患者の不本意およびまれなイベントのために治療は開始されなかった。 しかし,患者は部分てんかんとして現在進行中の発作を呈していた。 彼はめまいのオーラと診断された突然のめまいの再発感情を説明しました。 さらに、彼は(a)無反応とトルコの祈りの暗唱、(b)口頭自動、または(c)手をいじると音を発する—すべての部分的に強直間代発作に進化のいずれかを示し ビデオ脳波は現在、右と左の側頭葉の両方に局所スパイク波複合体を示した。 発作のセミオロジーにより両側側頭葉てんかんと診断し,レベチラセタムによる投薬を開始した。
31歳の時に発作が完全に抑制されなかったが、側頭葉てんかんのさらなる診断の一環として、患者はfdg-PETを受けた(図1)。 しかし、神経変性運動障害の疑いを提起したマークされた、非常に珍しい線条体代謝低下もありました。 その結果、広範なフォローアップ評価が開始されました(図2参照)。 Fp-CIT-SPECTは、黒質線条体の完全性の低下に沿って、線条体ドーパミントランスポーターの可用性の適度に両側の損失をもたらした行われました(図1)。 高分解能MRIでは、尾状核の両側萎縮が認められました(図3)。 神経学的検査では,離散的な束縛歩行パターンと運動低下を示したが,錐体外路運動系の他の愛情はなく,摂食ジストニアのような球症状はなかった。 神経伝導検査で感覚運動軸索多発神経障害が確認された下肢の低反射状態とは別に,神経学的検査は正常であった。 神経心理学的症状には不安障害の側面が含まれ、患者は短期間の記憶喪失を伴う困難を報告した。 しかし、神経心理学的検査では、明らかな記憶障害が確認されず、むしろ非特異的な気晴らしが確認され、その時点で不十分な発作抑制によってさらに強化されていた可能性がある。 実験室試験は徴候の癲癇(すなわち)のために否定的でした。、自己免疫性脳炎抗体、抗ニューロン抗体)。 脳脊髄液は炎症の徴候を示さなかったが、中等度のタンパク質増加(765mg/l)を示した。 脳脊髄液中の生体アミンの分析により,最近の発作に関連するグルタミンの上昇が明らかになった。 末梢血では、アカントサイトの0.4%が見出された。 ウィルソン病の血清学的検査は陰性であった。 顕著なのは、クレアチンキナーゼの持続的な上昇(範囲:1,000–3,000U/l)であり、ミトコンドリア障害の疑いのある診断につながった。 さらなる調査のために、正常な結果を示した虚血性乳酸試験を行った。 腓腹筋筋生検を行ったが,ミトコンドリア機能と呼吸鎖の解析は正常であった。
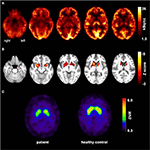
図1. 分子イメージング研究の結果。 横軸FDG-PET画像(A)は軽度の両側中側頭下代謝低下(中側頭下発作の起源に沿って)だけでなく,健常対照と比較して非常に有意であることが判明した著しい線条体代謝低下を示した。 追加のFP-CIT-SPECT検査(C)はまた、黒質線条体の完全性の低下を目の当たりにして、線条体ドーパミン輸送体の中等度の損失を明らかにした(年齢マッチした健康

図2. インデックス患者の臨床および診断経過。
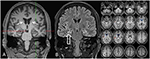
図3. MRIでは両側尾状頭萎縮と右側海馬硬化症を示す小さくて高強度の右側海馬を示した。 左の海馬は幾分hyperintenseですが、atrophicではありません。 両側尾状頭萎縮はCVR分析により確認され,青色は灰色の減少を示し,赤色は脳脊髄液量の増加を示した(C)。
家族歴は、彼自身が有意な病歴を持っていなかった彼の両親(最初のいとこ)の同族性を明らかにした。 彼の6人の兄弟のうち、2人(20-30歳の間)も今では全身発作に苦しんでいました(図4参照)。 彼の兄弟の医療記録は私たちには利用できませんでした。
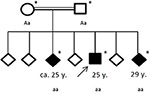
図4. 影響を受けた家族の血統、年の最初のてんかん発作の年齢(y)。 矢:索引のパテント。 アスタリスク:遺伝子検査が行われました。 Aa、ヘテロ接合;aa、ホモ接合c.4326T>A(P.Tyr1442*)。
遺伝性神経変性運動障害の仮説の下で、患者とその家族は遺伝子検査のために紹介されました。 エクソームシーケンシングは、三つの影響を受けた兄弟におけるChAc遺伝子VPS13Aにおけるホモ接合新規切り捨て変異c.4326T>A(P.Tyr1442*)を明らかにした。 同族の両親は両方ともヘテロ接合性であった。 エクソームシークエンシングにおいて他の変異体または変異は同定されなかった。
実際、33歳でのフォローアップでは、指数患者は依然として錐体外路症状しかなく、彼の姉妹には何も存在しませんでした。 右上肢のプライミング下でのみわずかな剛性と上肢の軽度の運動緩慢の印象を与えた。 レベチラセタムとラコサミドによる治療では,患者は発作を伴わなかった。
Discussion
本症例では、ChAcがvps13A遺伝子の新しい対応する突然変異のために重要な運動症状を欠いている側頭葉てんかんで臨床的に現れることを示
この疾患の経過は、典型的には進行性運動障害(舞踏病、ジストニア、パーキンソニズムを含む)、認知的および精神的変化、およびアカントサイトーシスおよび高血症の血清学的マーカーを伴う筋変性症状を特徴とする。 発作は以前にChAc(6)の症状として注目されてきたが、運動障害は依然として重要な症状と考えられている。 てんかん、より具体的には側頭葉に由来するてんかんは、ChAcの過小評価された表現型である可能性があるという証拠が生じる。 Peluso et al. 大脳基底核の関与を示す神経画像所見を提示しながら、46歳であっても、運動障害を示さなかった、私たちと同様の患者を報告した(7)。 それに応じて、Scheid e t a l. 主な症状として中側頭葉てんかんおよび硬化症(MRI研究に基づく)に苦しんでおり、ChAcと診断された三人の患者を記載した(8)。 さらに、Mente e t a l. 最近、大脳基底核萎縮だけでなく、海馬硬化症(9)だけでなく、表示されたChAc患者の剖検結果を提供しました。 我々のケースに沿って、海馬の両側の関与は、一般的な知見(であると思われる10、11)。 しかし、発作と硬化症との正確な関係は、依然として鶏または卵の問題である。 海馬の構造発達におけるコレインの相関役割はまだ完全には理解されていない。 興味深いことに、ChAc構造変化のマウスモデルでは、gaba(A)受容体ガンマ2とそのアンカリングタンパク質コレインの海馬タンパク質発現が増加した(12)とし
臨床ルーチンでは、この表現型は正しい診断を見つけるのに依然として困難である可能性があります。 我々の患者の神経変性疾患に対する最初の重要なヒントは、線条体グルコース代謝の顕著な低下とFDG-PETとFP-CIT-SPECT上の黒質線条体の完全性の中等度の低下で ケースシリーズの説明に基づいて、機能イメージング結果の小さなコレクションは、代謝とドーパミン作動性機能障害の変化は、主に尾状核と被殻(13)で発生す 脳MRIは、ChAc(14)の典型的な所見として記載されている疾患の経過中に尾状核の萎縮を明らかにした。 したがって、明確な神経学的特徴は、通常、舞踏病、摂食ジストニア、口腔顔面舌ジスキネジア、およびチックを含み、パーキンソニズムおよびジストニア(3)のような大脳基底核愛情の他の症状と一緒に含まれる。 私たちの患者で観察された線条体の変化は、病気が進行するにつれて最終的に発生する可能性のある症状(すなわち、前駆症状画像所見)を引き起こすしきい値を下回っていたことを推測することは魅力的である。 したがって、我々は、この孤児病の敏感な診断ツールとして構造および分子イメージングを奨励します。
ChAcには、表皮細胞数が5~50%の記述があります(3)が、表皮細胞が疾患の経過の後半にしか現れないことを示す症例報告がいくつかあります(15)、非常に低 したがって、アカントサイトは診断を行うために必須とみなされるべきではない。 さらに、てんかんはChAcの他の典型的な症状を隠すので、診断を遅らせる可能性があります。 特に、非常に一般的な人格変化や認知悪化などの精神症状は、誤って解釈され、薬物関連の副作用(すなわち、レベチラセタム)または発作関連に起因する可能性がある。 二次性高血症は、一般的な強直間代発作の後に見られ、持続的な上昇が見落とされる可能性がある。
我々のケースはまた、特にまれな疾患や症状が曖昧な場合に、正しい診断を確立するためのエクソームシーケンシングの重要な利点を強調しています。 哺乳類のVPS13(A–D)遺伝子のファミリーが注目されており、常染色体劣性パーキンソン病や常染色体劣性脊髄小脳失調症などの他の神経学的疾患で変異が同定されている(17)。 VPS13Dの劣性突然変異は、小児発症運動障害を引き起こす(18)。 ChAcの根底にある病態生理はまだ完全に解読されていませんが、VPS13Aは、脳および他の組織の膨大な数で遍在的に発現されているタンパク質コレインをコードすることが示された(19)。 研究は、それが細胞骨格アーキテクチャ、エキソサイトーシス、および細胞生存(を調節するシグナル伝達経路に関与していることを明らかにする20)。 ChAcを有する9人の患者において発作支配表現型を示すことが記載されている別の変異は、VPS13A遺伝子におけるc.2343del変異である(21)。 同じ遺伝子の特定の突然変異は、例えば脳の異なる領域に影響を与えることによって、独特の表現型を引き起こすコレインのある種の異常をもたら しかし、根底にある病態生理学は依然として投機的であり、原因となる突然変異のさらなる文書化は興味深いものになるであろう。 我々は、前に記載されていない切り捨て変異c.4326T>A(P.Tyr1442*)は、すべての影響を受けた家族のメンバーで優勢なてんかんと同様の表現型をもたらすこ
おわりに
てんかん(特に両側側頭葉てんかん)はChAcの優勢な表現型である可能性が高い。 したがって、追加の赤旗(高血症、家族歴、多発性神経障害など)の検索を慎重に行う必要があります。 これらのツールが広く利用可能であるとして、疑わしい場合には、血液や脳MRI中のアカントサイトの検索を追加する必要があります。 疑わしい場合は、線条体の関与を検出するのに敏感であるため、fdg-PETおよび/またはFP-CIT-SPECTで診断を補完する必要があります。 神経変性疾患の指標所見を有する常染色体劣性てんかんでは、vps13A単一遺伝子検査またはchorein Western blotを使用して正しい診断を確立する必要があります。 後者は、遺伝的に定義された病因のない他の神経変性疾患においても考慮されるべきである。
倫理声明
この研究への参加について、患者から書面によるインフォームドコンセントが得られました。 本症例報告の公表については,患者から書面によるインフォームドコンセントを得た。
著者の貢献
JW、MR、SKが患者管理に参加しました。 JWは原稿の最初の草案を書いた。 LF、HU、およびPMは図を準備しました。 すべての著者は原稿の改訂に貢献し、提出されたバージョンを読んで承認しました。
利益相反声明
HUはフライブルク大学医療センターのスピンオフであるVeobrain Gmbhの株主である。
残りの著者らは、この研究は利益相反の可能性があると解釈される可能性のある商業的または財政的関係がない場合に行われたと宣言している。
1. Walterfang M,Evans A,Leong Looi JC,Jung HH,Danek A,Walker RH,et al. ニューロカントサイトーシス症候群の神経精神医学。 Neurosci Biobehav Rev.(2 0 1 1)3 5:1 2 7 5−8 3. ドイ:10.1016/j.neubiorev.2011.01.001
PubMed要約/CrossRef全文/Google Scholar
2. ウォーカー RH. 神経組織細胞症症候群の管理。 振戦その他Hyperkinet. Mov。 (2015) 5:346. doi:10.7916/D8W66K48
PubMed要約/CrossRef全文/Google Scholar
3. Velayos Baeza A,Dobson-Stone C,Rampoldi L,Bader B,Walker RH,Danek A,et al. 舞踏病-アカントサイトーシス。 ジェネレビューズ®。 ワシントン州シアトル:ワシントン大学(1993年)。
4. Critchley EM,Clark DB,Wikler A. ベタリポプロテイン血症のないアカントサイトーシスおよび神経学的障害。 アーチノロール (1968) 18:134–40.
5. レヴァインIMエステスJWルーニー JM アカントサイトーシスを伴う遺伝性神経疾患。 新しい症候群。 アーチノロール (1968) 19:403–9.
PubMed Abstract/Google Scholar
6. Hardie RJ,Pullon HW,Harding AE,Owen JS,Pires M,Daniels GL,et al. ニューロアカントサイトーシス。 19例の臨床的、血液学的および病理学的研究。 Brain(1 9 9 1)1 1 4(Pt1A):1 3−4 9.
PubMed Abstract/Google Scholar
7. Peluso S,Bilo L,Esposito M,Antenora A,Rosa ADR,Pappata S,et al. 舞踏病-舞踏病のないアカントサイトーシス:臨床表現型を拡大する。 パーキンソン病関連 (2017) 41:124–6. 土井:10.2016/j.parkreldis.2017.05.013
PubMed要約/CrossRef全文/Google Scholar
8. Scheid R、Bader B、Ott DV、Merkenschlager A、Danek A.舞踏病-アカントサイトーシスにおける側頭葉てんかんの発症。 神経学(2 0 0 9)7 3:1 4 1 9−2 2. 土井:10.1212/WNL.0b013e3181bd80d4
PubMed要約/CrossRef全文/Google Scholar
9. Mente K,Kim SA,Grunseich C,Hefti MM,Crary JF,Danek A,et al. 舞踏病-アカントサイトーシスにおける海馬硬化症および側頭葉てんかん:臨床的、病理学的および遺伝的評価を伴う症例。 ニューロパソロールアップルニューロビオール… (2017) 43:542–6. 土井:10.1111/ナン。12403
PubMed要約/CrossRef全文/Google Scholar
10. Bader B,Vollmar C,Ackl N,Ebert A,la Fougère C,Noachtar S,et al. 舞踏病-アカントサイトーシスにおける頭蓋内脳波で両側側頭葉てんかんが確認された。 (2011)20:340-2. 土井:10.1016/j.発作。2010.12.007
PubMed抄録/CrossRef全文/Google Scholar
11. Marson AM,Bucciantini E,Gentile E,Geda C.Neuroacanthocytosis:イタリアの家族における臨床的、放射線学的、および神経生理学的所見。 ニューロルサイ… (2003) 24:188–9. 土井:10.1007/s10072-003-0123-1
PubMed Abstract|CrossRef全文|Google Scholar
12. 倉野Y,中村M,市場M,松田M,水野E,加藤M,et al. コレイン欠乏症は、ゲフィリンとGABA(A)受容体のアップレギュレーションにつながります。 Biochem Biophys Res Commun. (2006) 351:438–42. 土井:10.1016/j.bbrc。2006.10.070
Pubmed要約/CrossRef全文/Google Scholar
13. Ehrlich DJ,Walker RH. 機能的神経イメージングと舞踏病:体系的なレビュー。 J-Clin Movied。 (2017) 4:8. 10.1186/40734-017-0056-0
PubMed Abstract|CrossRef全文|Google Scholar
14. コノリー BS、ハズラティL-N、ラングAE。 舞踏病-アカントサイトーシスにおける神経病理学的所見:パーキンソニズムと発作の根底にあるメカニズムへの新しい洞察。 アクタニューロパトール (2014) 127:613–5. ドイ:10.1007/s00401-013-1241-3
PubMed Abstract|CrossRef全文|Google Scholar
15. Sorrentino G,De Renzo A,Miniello S,Nori O,Bonavita V.舞踏病-アカントサイトーシスの過程におけるアカントサイトの後期出現。 Jニューロルサイ… (1999) 163:175–8.
PubMed Abstract/Google Scholar
16. バイロイトC、ボルグM、フェレーロ-ヴァッヒャー C、シャウセノー A、レブルンC。 Choréo-acanthocytose sans acanthocytes。 レヴュー-ニューロル… (2010) 166:100–3. 土井:10.1016/j.neurol.2009.03.005
クロスリファレンス全文/Google Scholar
17. Lesage S,Drouet V,Majounie E,Deramecourt V,Jacoupy M,Nicolas A,et al. 常染色体劣性パーキンソニズムにおけるVPS13C機能の損失は、ミトコンドリア機能不全を引き起こし、PINK1/パーキン依存性mitophagyを増加させます。 Am J Hum Genet. (2016) 98:500–13. 土井:10.1016/j.ajhg.2016.01.014
PubMed要約/CrossRef全文/Google Scholar
18. Gauthier J,Meijer IA,Lessel D,Mencacci NE,Krainc D,Hempel M,et al. >VPS13Dの劣性変異は、小児発症運動障害を引き起こす。 アン-ノイロール (2018)83:I6. 土井:10.1002/ana.25204
PubMed要約/CrossRef全文/Google Scholar
19. 倉野Y,中村M,市場M,松田M,水野E,加藤M,et al. In vivoでの分布とコレインの局在。 Biochem Biophys Res Commun. (2007) 353:431–5. 土井:10.1016/j.bbrc。2006.12.059
Pubmed要約/CrossRef全文/Google Scholar
20. Lang F,Pelzl L,Schöls L,Hermann A,Föller M,Schäffer TE,et al. ニューロン、赤血球およびそれ以降-コレインの多様な機能。 Neurosignals(2 0 1 7)2 5:1 1 7−2 6. 土井: 10.1159/000485457
PubMed Abstract|CrossRef全文|Google Scholar
21. Benninger F,Afawi Z,Korczyn AD,Oliver KL,Pendziwiat M,Nakamura M,et al. C.2343del VPS13A遺伝子変異を伴う舞踏病-アカントサイトーシスにおける提示および顕著な症状としての発作。 ら(2 0 1 6)5 7:5 4 9−5 6. 土井:10.1111/epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar