mucopolysaccharidoses:early diagnostic signs in infants and children
Mucopolysaccharidoses(MPS)は、グリコサミノグリカン(Gag)の分解に必要なリソソーム酵素の欠乏によって引き起こされる臨床的に異質な疾患群である。
MPSは進行性および全身性の臨床症状を特徴とする。 それらの生化学的および遺伝的異質性にもかかわらず、異なるタイプは、剛性(弛緩があるMPS IVを除く)と痛み、粗い顔の特徴、角膜混濁、鼠径部または腹部ヘルニア、再発上気道感染症、心臓弁疾患、手根管症候群、および可変神経学的関与を伴う関節および骨格異形成を含む様々な組み合わせにおいて重要な臨床的特徴を共有する。 これらの特徴は、通常、重度の形態のための人生の最初の数ヶ月で、より弱毒化された形態のための幼児期に現れるが、しばしば過小評価され、一般的に
これらの疾患の希少性および臨床的提示の変動性は、しばしば診断遅延をもたらす;これは、重度の形態では数ヶ月から、弱毒の形態では数年までの範囲である(Rigoldi et al. この補足では)。 それにもかかわらず、重度のプレゼンテーションへの介入のための迅速な進行と緊急の必要性のために、診断の遅れの数ヶ月でさえ、患者の将来の健康
私たちの目的は、このレビューで小児科医に初登場を警告するべき重度の形態の非常に初期の兆候を強調することです。
重度のMPSを持つ子供にはどのような徴候/症状が期待できますか?
様々な形態のMPSを示唆する症状および徴候を年齢別に表1に報告している。 人生の最初の6ヶ月では、鼠径ヘルニア、異常に頻繁な呼吸器感染症、耳炎、および臓器肥大は、特に最も早熟な重度の発症を有するMPS I(ハーラー症候群)で、MPS患者 さらに、6ヶ月から12ヶ月まで、これらの乳児は非常に頻繁にギブス(トラコ-腰椎後弯症)、心雑音、臍ヘルニア、軽度の低血圧、および成長遅延を発症する。 彼らはまた、典型的な粗い顔の外観を有することができる(図。 1). MPS II、VI、およびVIIは、同様の初期表現型を提示することができます。 MPS IV患者は、最初の12-18ヶ月で人生の最初の数ヶ月で股関節形成異常の出現を伴う初期の骨格表現型を有し、ハト胸(pectus carinatum)を有する。 MPS IIの幼児は同じような介入としかしより遅い手始めと示します。 人生の二年目に、MPS I Hurler症候群の乳児は認知遅延を発症し、MPS II患者は過増殖、頻繁な気道感染症、および複数の手術のためにのみ同定されるかもしれない。 何人かのMPS IIの患者はまた18そして24か月間の多動を開発します。 人生の三年目には、多動および認知遅延は、MPS IIおよびMPS IIIで容易に認識可能である.
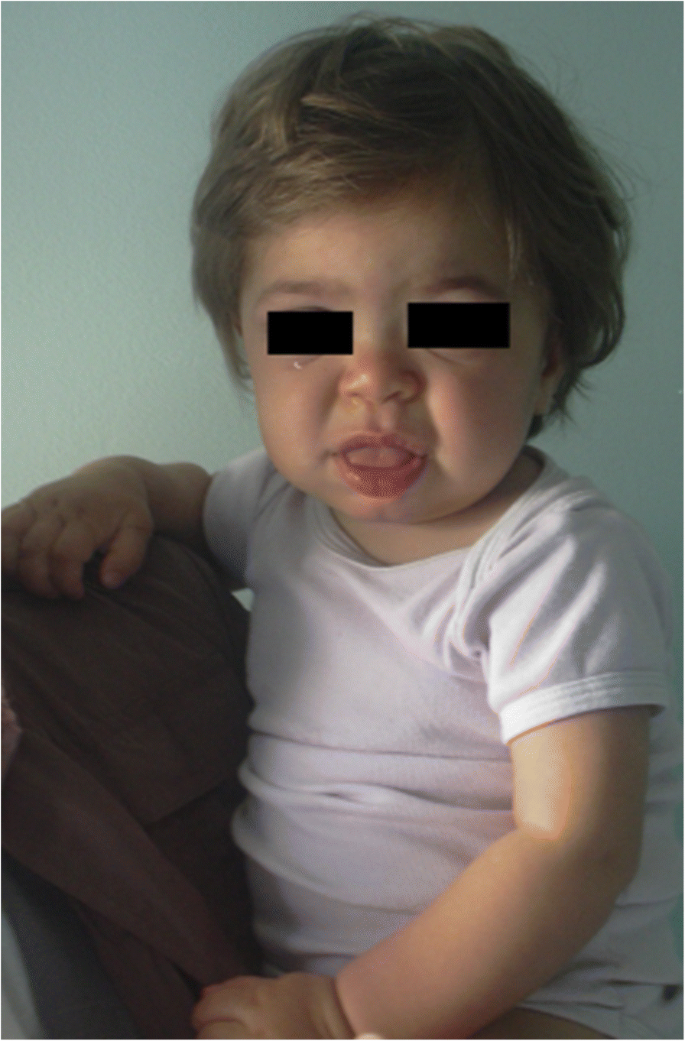
1歳の患者のMPS IH。 粗い顔:平らな鼻橋、macroglossia、前頭bossing
要約すると、骨格症状は典型的であり、時には早熟である。 粗相および認知遅延は早期の顕著な徴候ではない。
様々なタイプの国会議員の異なるプレゼンテーションは何ですか?
体細胞および認知関与を有するMPS
ムコ多糖症I型(MPS I;OMIM#252800)(図。 1、2、3、4および5)は2つのGAGsのmultisystemの蓄積をもたらすlysosomal hydrolase α-l-iduronidaseの不足によって引き起こされます、:dermatanの硫酸塩(DS)およびヘパランの硫酸塩(HS)。 その重症度に基づいて、MPS Iは伝統的に3つの臨床表現型に分けられていますが、これらの形態は疾患の重症度の連続を表しています。 最も重篤な形態であるHurler症候群は、典型的には人生の最初の年の間に提示される。 影響を受けた子供は急速に処置の不在の最初の十年以内の死に導く多臓器系の重要な認識減損そして体細胞の病気を開発します。 Hurler−Scheie症候群およびScheie症候群として知られるmps iの弱毒型は、それぞれ、症状の後発、より長い平均余命、および軽度または中枢神経系(CNS)の関与を特徴とする。 遺伝は常染色体劣性であり、少なくとも50%の症例では遺伝子型に基づいて表現型予測が可能であり、残りの50%では新たな突然変異が検出される。 有病率はおよそ100,000の生きている生れに付き1です。

MPS IH. 9ヶ月の患者のギブス。 b脊椎3歳の患者の脊椎のx線。 重要な脊柱後弯症および脊柱管狭窄症を示す脊柱のC T2重み付けされた磁気共鳴のイメージ

MPS IH. Dysostosis multiplexのX線証拠。 3歳の患者の肋骨の肥厚および18ヶ月の別の患者のb股関節形成異常

MPS IH:18ヶ月の患者の爪の手;b手のX線。

MPS IH. 頭蓋骨の皮質骨の肥厚およびsella turcicaの異常な”J字型”立体配座。 B FLAIRおよびc T-2重み付け磁気共鳴画像における脳室周囲空間の拡張17ヶ月の患者の
ムコ多糖症II型(MPS II;OMIM#3 0 9 9 0 0)(図3)。 6)、別名ハンターシンドロームは、DSおよびHSの必然的なGAGの蓄積を用いる酵素のiduronate-2スルファターゼ(I2S)の不足の結果です。 有病率は140,000–156,000人の男性の生きている生れに付きおよそ1です。 特に、これは唯一のXリンクされたMPSであり、女性のキャリアは通常無症候性です;但し、X染色体、homozygosity、または歪んだX不活性化の異常が例外的に影響を受 表現型の表現はまた臨床重大度の広いスペクトルに及ぶ。 重度の症例では、著しい進行性の神経学的関与と体性疾患が共存し、死は通常、人生の第二十年に起こります。 神経学的機能障害が最小またはまったくない他の患者は重度の体細胞関与を示すことがあり、スペクトルの反対側には、成人期に生き残る最小限の体細胞症状および正常な知性のみを有する患者がいる。 初期の徴候および症状は、重篤な形態のMPS IおよびIIによって共有される.

MPS II. 軽度の特徴的な顔の特徴のみを持つ3歳の患者
胎児水腫の散発的な症例は、一般に、これらの子供は出生時に正常に見えるが、MPS Iに記載されている。 MPS IIのものは、通常、少し後に発生しながら、MPSの最初の兆候は、私は通常、人生の非常に最初の数ヶ月の間に表示されます。 Kiely et al. 55人のHurler患者を対象としたレトロスペクティブ研究では、呼吸器症状、摂食困難、鼠径ヘルニア、中耳炎が6ヶ月以上の中央値で出現し、後弯症、心臓異常、頭囲の拡大、低血圧、器官肥大、角膜混濁、関節制限、臍ヘルニアが6-12ヶ月の間に出現したことが観察された。 臍ヘルニアは、MPS IおよびIIにおける最も初期の提示機能の一つであり、鼠径ヘルニアは、患者の約60%で報告されている。
MPS IおよびIIのもう1つの一般的な初期の手がかりは、分泌物が増加した上気道再発性感染症である可能性があります。 明らかな伝染のない頻繁な中耳炎、慢性の再発鼻炎および耐久性がある鼻汁は特定ではないですが、診断疑いの増強で特定の印と関連付けられたら助け 口腔咽頭内のGAGの貯蔵は、扁桃腺およびアデノイドの拡大をもたらし、上気道合併症に寄与する。 別の頻繁な発見は、特に夜間の騒々しい呼吸であり、後の段階で閉塞性睡眠時無呼吸と関連している。 中耳の耳小骨の頻繁な中耳感染症およびdysostosisの結果として、難聴は頻繁に起こり、伝導性および感音性の両方の欠損を伴う。 気管気管支軟化症は一般的に観察され、急性気道閉塞または虚脱につながる可能性があり、これはその後の数年間の主な死因の1つである。
アデノイド切除術、扁桃摘出術、鼓膜吻合術は、ヘルニア修復とともに、MPS IおよびII患者のより頻繁かつ早期の外科的介入であり、診断を知る前に(症例の50%以上で)行われることが多い。
再発性下痢はMPS Iの初期段階では非常に一般的であり、神経学的関与を有するMPS II患者にも存在する可能性があるが、軽度の罹患患者では珍しい。
ギブス変形(背側腰椎後弯)(Fig. 2)は8の中央値年齢で臨床的に明白になります。MPS Iでは6ヶ月から1年で、一般的にMPSの非常に具体的で早熟なマーカーを表しています。 椎骨体は平らになり、びっしりとしているように見え、脊髄神経捕捉、急性脊髄損傷、大西洋後頭不安定性などの後の合併症を引き起こす可能性があ 1歳の後で、共同拘縮およびgenuのvalgumはMPSによって影響されるすべての幼児の早いより2歳で容易に検出可能である共同減損および進歩的な骨格異形成 これらの骨格症状はすべて、MPSの典型的なものであり、dysostosis multiplexと命名されています。 肋骨の肥厚は、x線写真で見ることができる(Fig. 3a)、長い骨は広いシャフトと短く、膝は外反および内反の醜状に傾向があります。 指節異常症および滑膜肥厚は、特徴的な爪の手の変形をもたらす(図10)。 4)、病気の疑いを上げるかもしれないもう一つの典型的な特徴である。 情報通の異形成は頻繁にこれらの子供が整形外科の専門家によって最初に見られるかもしれない理由です。 典型的には、骨盤は不十分に形成され、大腿骨頭は小さく、coxa valgaは一般的であり、進行性および衰弱性の股関節変形を伴う(図10)。 3b)。
MPS IとMPS IIの間には顕著な違いがあり、mps Iでは骨格の成長は3歳で減速し、MPS II患者では最初の5〜6歳で過成長を示し、その後の成長が遅くな
口腔領域および顔面骨の軟部組織におけるギャグの蓄積による顔の特徴(鼻孔がフレアした広い鼻、眼窩上隆起、大きな丸い頬、厚い唇、突出舌の拡大)の粗大化は、最初の2年以内に明らかになり、ハーラー病では中央値年齢1.1歳、ハンター病の重度の形態では18ヶ月から4年の間に明らかになるが、微妙な表現型の変化は早期に検出することができる(最初の後半には早くも検出することができる)。年)特定の経験を持つ小児科医による(図。 1). 大頭脳症は徐々により明白になり、舟状脳症が一般的である(図10)。 5)、しかし小頭症はHurlerの患者でまた可能です。 顔と体の多毛症は、常に24ヶ月の年齢で明らかです。 皮膚は肥厚し、非弾性である可能性があります。 特に、何人かのMPS IIの患者はハンターシンドロームのpathognomonic上腕の上部の背部そして側面の象牙白い丘疹(小石そっくり)として記述されている特有な皮膚病変を
Mps I患者における心筋症および弁小葉の進行性の肥厚および硬化は、僧帽弁およびより少ない頻度で大動脈逆流を引き起こす可能性がある。 雑音は、人生の最初の数ヶ月で検出されることがあり、時には病気の最初の兆候として報告されています。 心臓の関与は、MPS IIのほぼすべての患者にも存在するが、これは約5歳から始まる。 弁疾患は、右および左心室肥大および心不全につながる可能性があります。
進行性肝脾腫によって引き起こされる腹部の隆起は、生後6ヶ月後に容易に明らかであるが、肝臓および脾臓にGAGsを貯蔵しても臓器機能不全には
角膜混濁は、生後15ヶ月より早いMPS Iのほぼすべての個体で発生し、重度の視覚障害を引き起こす可能性があります。 逆に,角膜混濁はハンター症候群の典型的な特徴ではないが,スリットランプ検査では視力に影響を与えない離散的な角膜病変を明らかにする可能性がある。 緑内障は一般的な所見ではないが、網膜機能障害が検出される可能性がある。
初期の精神運動発達は一般的に正常と認められていますが、MPS Iでは通常18ヶ月齢までに発達遅延が明らかになり、進行性の精神的低下が重度の知的障害につながります。 これらの子供たちの言語能力は非常に限られています。 MPS IIの開発マイルストーンの世界的な遅延は、2年以降から明らかになります。 しかし、重度のハンター病の主要な神経学的特徴は、典型的には弱毒表現型を有するものでは観察されない、多動、頑固さ、および積極性などの顕著な行動
ムコ多糖症VII型(MPS VII;OMIM#253220)は、スライ症候群としても知られており、β-グルクロニダーゼの活性が不足し、コンドロイチン硫酸(CS)、DS、HSのリソソーム貯蔵があり、細胞および器官の機能不全につながる超まれなMPSである。 最初の患者はSlyらによって記載された。 1973年には合計145人の患者がこれまでに文献に報告されている。
MPS VIIの臨床的提示と疾患進行は、初期、重度、多系統症状および最初の数ヶ月での死亡から、後に発症する軽度の表現型、正常または正常に近い知性、およ
MPS VII患者の表現型特性は、MPS IおよびMPS II(低身長、骨格異形成、大頭症、歯肉肥大、再発性耳感染、肝脾腫、ヘルニア、心臓関与、肺機能の低下、認知障害)に似ている。 記載された患者の予想外に高い割合(41%)は、非免疫新生児胎児水腫(NIHF)の病歴を有する。 これらの患者の病気の出生前の手始めにもかかわらず、13のうち23は彼らの遅い十代の若者たちに中間コースに穏やかの幼年時代を生き残った。 したがって、NIHFの存在は、患者が幼児期に生存している場合、それ自体で臨床経過の可能性のある重症度を予測するものではない。 もう一つの初期の徴候は大頭蓋症であり、心臓弁の関与および上気道および下気道の反復感染は人生の最初の2年間に現れる。 中等度の精神遅滞および言語障害を伴う進行性難聴も時間とともに明らかである。
要約すると、重篤な形態のMPS私は非常に早期のマルチシステム発症を持っています(生後6ヶ月以内); MPS IIは類似しているが、後の発症を伴い、MPS VIIは出生前の発症を有し、頻繁なNIHFおよび上気道および下気道の早期重度の関与を有する。
主に神経学的および認知的関与を有するMPS
ムコ多糖症III型(Mps III、サンフィリッポ症候群としても知られている。 7)は、一般的な神経学的提示を持っています。 MPS IIIは、HSの異化に関与する四つの酵素のいずれかの欠乏によって引き起こされるリソソーム貯蔵障害である。 4つの異なるサブタイプが知られている:MPS IIIタイプA(OMIM#2 5 2 9 0 0)、タイプB(OMIM#2 5 2 9 2 0)、タイプC(OMIM#2 5 2 9 3 0)、およびタイプD(OMIM#2 5 2 9 4 0):それぞれ異なる酵素欠乏症に起因すhgsnat遺伝子;D型,N—ACETILGLUCOSAMINA—6-solfato SULFATASI/gns遺伝子)。 すべての4つのタイプ(AからD)は常染色体劣性遺伝を持っています。 MPS IIIは、0.3と4の間の推定有病率を持つMPSの中で最も頻繁です。サブタイプとレースに応じて、すべての100,000新生児のための1ケース。

MPS IIIA. A2.5とb5歳の同じ患者。 神経学的障害の進行を示す異なる顔の表情に注意してください
Sanfilippo症候群の患者は、人生の第二および第三の年に進行性の認知障害を示す。 MPSの大部分とは対照的に、体細胞の特徴は比較的明らかではなく、CNSの関与は顕著である。 スピーチの開発の遅れは通常親によって気づかれる最初の印であるが、主要な心配は進歩的な精神低下に先行している行動の問題(多動、心配し、積極的な行動、睡眠の妨害)である場合もある。 これらの症状は、行動障害、注意欠陥多動性障害(ADHD)、および自閉症スペクトラム障害として誤診を引き起こす可能性があります。 てんかんも存在する可能性があります。
睡眠障害は、その発生率が80-90%まで報告されているのが一般的な特徴です。 彼らは眠りに落ちることの困難と頻繁な夜間の覚醒からなり、一部の患者では昼夜のリズムが完全に逆転する。
これらの症状はすべて管理が非常に困難であり、家族全員の生活の質に大きな心理的影響を与えます。
一般に、表現型は4つのサブタイプで非常に類似している可能性がありますが、Sanfilippo BとCの臨床経過はそれほど深刻ではない表現型を特徴としてい
非神経学的症状は、通常、MPS IIIでは他のMPSよりも顕著ではない。 再発性の耳、鼻、および喉の感染症は、より若い年齢で観察される。 再発性中耳炎、中耳耳小骨欠損、および内耳の異常は難聴を引き起こす可能性があります。 過剰な厚い分泌物および解剖学的変化は、気道の閉塞を引き起こす可能性がある。 人生の最初の数年間では、下痢が頻繁に報告されますが、便秘は高齢の患者でより一般的です。
身体検査では、粗い顔の特徴を示すことができますが、これらはあまり顕著ではありません。 患者は大頭症、中間の急に燃え上がることおよびsynophrysの広い眉毛があります、毛は通常乾燥し、粗く、hypertrichosisは共通です。 若年患者では、軽度の肝腫大および臍ヘルニアおよび鼠径ヘルニアが頻繁に観察される。 まれで、しばしば軽度の拘縮は、主に肘に見られる。 成長は12年より古い患者のおよそ半分で影響を受けます。
要約すると、MPS IIIまたはSanfilippo病は非常に軽度の体細胞徴候を伴う顕著な神経学的関与を有する。 多動、ADHD、および攻撃的行動を発症する2〜3歳の幼児は、mps IIIが疑われる可能性があります。
体細胞関与が多いMPS
mps IVおよびmps VIは体細胞関与 これらは、知的能力を惜しまず、主に骨格(MPS IVA)、または身体のすべての器官/系(MPS VI)に影響を与える進行性の状態である。 遺伝は常染色体劣性です。 症状が悪化する速度は、影響を受けた個人によって異なります。
ムコ多糖症IV(Mps IV、モルキオ症候群としても知られている。 8)は、N-アセチルガラクトサミン-6-スルファターゼ(GALNS遺伝子)の欠乏によるMPS IVA(Morquio A症候群;OMIM#253000)、ケラタン硫酸塩(KS)およびCSの蓄積によるmps IVB(Morquio B症候群;OMIM#253010)、GM1患者のように尿KSおよびオリゴ糖排泄につながるベータガラクトシダーゼ活性の欠乏によるMPS IVB(Morquio B症候群;OMIM#253010)である。 MPS IVBは確かにGM1-ガングリオシドーシスの様々な形態に対立遺伝子であるが、GM1ガングリオシドーシスで見られる精神運動悪化を欠いている。

イヴァ議員 典型的な”前方のbeaking”の面の”かい型の”肋骨そして平らにされ、円形にされた椎骨を示すbのantero後部およびcの側面x線を持つ4歳の患者
検出可能な酵素活性のない重度のMPS IVA患者は、通常、生後1年の間に最初の症状を明らかにするが、4-5歳の前に診断されることはめったにない。 Morquio A患者の自然史に関する情報は、主に二つの幅広いデータコレクションから派生しています。 減少した成長率は約18ヶ月齢で始まり、成長は約7または8歳で停止します。 他のMPSとは対照的に、Morquio a関節は、通常、緩いと非常に柔軟性(hypermobile)ですが、いくつかの関節(通常は腰と肩)は、運動の制限された範囲を有することができます。 このような骨格の関与を有する患者は、脊椎骨端異形成、偽軟骨異形成、多発性骨端異形成、または両側脚-カルベ-ペルテス病の診断を受けるか、または評価を受けることができる。
一定の特徴は歯類形成不全である; したがって、大西洋後頭関節の脱臼は、子宮頸部の安定化が早期に行われない場合、麻痺または死に至る、球および脊髄への圧迫および損傷を引き起こ
MPS IVB患者の臨床症状は非常に似ていますが、低身長はあまり顕著ではありません。 彼らは軽度の粗い顔の特徴、難聴、角膜混濁、弁膜性心疾患、および頻繁な上気道感染症を持っています。 それらはまた鼠径ヘルニアおよび穏やかなhepatomegalyと示すかもしれません。 骨格異形成の特徴には、板状突起、可能性のある子宮頸部亜脱臼を伴う歯状形成不全、kypho-脊柱側弯症、coxa valga、および狭窄した腸骨翼が含まれる。<1 8 4 1><4 7 1 9>ムコ多糖症VI(Mps VI、Maroteaux−Lamy症候群としても知られている;OMIM#2 5 3 2 0 0;図1 4)。 9)は、第5染色体上に位置するアリールスルファターゼB(ARSB)遺伝子の病原性変異によるものである。 結果として、酵素アリールスルファターゼB(ASB)(N-アセチルガラクトサミン4-スルファターゼ)の活性の低下または不在は、その細胞蓄積を決定するDSの分解を損

明らかに粗い顔と骨格dysostosisを持つ2歳の男の子
検出可能な酵素活性のない患者は、重度の形態のMPS VIを有し、幼児期に症状を示す。 彼らの体細胞表現型はHurler症候群に似ています。 ほとんどの場合、2歳または3歳までに、重度で進行性の骨格損傷、dysostosis multiplexと呼ばれることが明らかになります。 この骨格異形成には、異形成性大腿骨頭、異常な椎体、不規則な鎖骨、低形成性遠位尺骨および橈骨、および異形成性および短い中手骨を伴う股関節異形成が含まれ、手根骨および足根骨は低形成性であり、不規則なプロファイルを有する。 成長速度は、多くの場合、最終的な高さは、一般的に120センチメートルよりも低いと、人生の最初の年後に遅くなります。 他の国会議員と同様に、初期の兆候は粗い顔の特徴です。 さらに、患者は、典型的な突出した腹部、肝腫大、臍および/または鼠径ヘルニア、および初期の乳児期における心臓の関与を有する。 一次知的障害は存在しないが、結果として頭蓋内圧亢進症および乳頭浮腫を有する一部の患者では水頭症が見られる。 Pectus carinatum、堅く、引き締まった接合箇所、脊柱側弯症および脊柱後弯症および頚部脊髄の圧縮は時間と悪化します。
要約すると、MPS IVおよびVIはCNSの関与を示していない。 Mps IVは関節の弛緩を示すユニークなMPSであり、重度のMPS VI発症はMPS Iで観察されたものと同様である