Artikkel
Josivan Gomes Lima1*, Marcel Cat Hryvo Ferreira dos Santos1, Julliane Tamara Araújo De Melo Campos2
1departamento de medicina cl@nica, endokrinologi og metabologia disiplin. Sykehus Universitetá Onofre Lopes, Universidade Federal do Rio Grande do Norte (UFRN), Natal, Rn, Brasil
2fakultet For Helsefag Ved Trairi, Federal University Of Rio Grande do North (UFRN), Natal, Rn, Brasil
Abstrakt
Medfødt Generalisert Lipodystrofi (CGL) er en sjelden og alvorlig autosomal recessiv sykdom. Pasienter er defekte i lagring av kroppsfett, og følgelig legger de fett i ektopiske vev, hovedsakelig lever, og kan utvikle skrumplever. Insulinresistens er et typisk funn som forårsaker diabetes som krever høye daglige doser insulin. I Delstaten Rio Grande do Norte, Brasil, har vi en av de største kohortene av pasienter med CGL. I denne artikkelen vurderer vi patofysiologi, klinisk bilde og behandling av denne sykdommen.
Innledning
Type 2 diabetes er et helseproblem i verden, og skyldes vanligvis overvekt og økt visceralt fett som forårsaker perifer insulinresistens og manglende evne i bukspyttkjertelen til å frigjøre insulin for å kompensere for denne motstanden. Andre mindre vanlige typer diabetes oppstår på grunn av spesifikke genetiske mutasjoner, som Medfødt Generalisert Lipodystrofi (CGL), også kjent Som Berardinelli-Seip Medfødt Lipodystrofi (BSCL). CGL er en autosomal recessiv sykdom som er klassifisert i fire typer, basert på genmutasjon. De endrede genene spiller viktige funksjoner for adipocyttdannelse, lipidproduksjon og riktig lagring inne i adipocyten. Mutasjonene reduserer fettvev med påfølgende avsetning av fett i ektopiske områder, forårsaker fettlever, endret karbohydratmetabolisme, alvorlig insulinresistens med hyperinsulinemi og akromegaloide egenskaper og dyslipidemi1-3. CGL syndrom har rundt 500 tilfeller rapportert i verden. I Brasil, I Delstaten Rio Grande do Norte (RN), har vi diagnostisert, behandlet og fulgt 54 tilfeller de siste 20 årene4, 5. I en deskriptiv studie med sekundære data estimerte vi totalt 103 pasienter i RN6. Dette indikerer en mye høyere prevalens enn det som er rapportert i litteraturen (1: 1 million) 7.
Triacylglycerol dannelse og lagring i lipiddråper
biosyntesen av triglyserider og fosfolipider (Figur 1a) starter med glycerol-3-fosfat acyltransferase (GPAT) acylating glycerol-3-fosfat i posisjon 1, danner 1-Acylglycerol-3-fosfat (lysofosfatidinsyre). Det følges av et annet acyleringstrinn i posisjon to av enzymet AGPAT (1-Acylglycerol-3-fosfatacyltransferase), som stammer fra 1,2-Diacylglycerol-3-fosfat (fosfatidinsyre). Det er et viktig mellomliggende trinn i biosynteseveien til både triglyserider og fosfoglyserider. DET er 11 isoformer AV agpat enzymer, kodet av forskjellige gener4. AGPAT1 OG AGPAT2 er de mest omfattende studerte. AGPAT1 er tilstede ved høye nivåer i testis, bukspyttkjertel og i mindre grad i fettvev og annet vev som hjerte, placenta, hjerne, lunge, MENS AGPAT2 er rikelig i fettvev. I de følgende trinnene oppstår det cytosoliske enzymet fosfatidinsyrefosfatase (PAP eller lipin) 1,2-diacylglycerol, og 1,2-diacylglycerolacyltransferase (DGAT) danner triacylglycerol4. Fosfatidinsyre og diacylglycerol kan også stamme fra andre fosfolipider som kardiolipin, fosfatidylinositol og fosfatidylkolin.
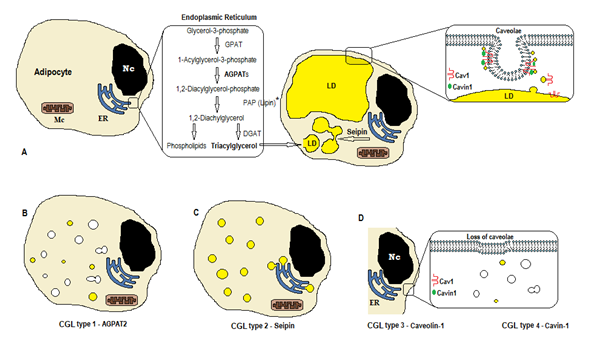
Figur 1. Skjema for triglyseridsyntese i HENHOLD TIL CGL-typer. (A) Normal syntese og lagring av triacylglycerol (TAG) i adipocyt. (B) Mutasjon AV AGPAT2 reduserer TAG produksjon (noen er fortsatt syntetisert under stimulering av Andre AGPATs). (C) Mutasjon av seipin genet redusere TAG syntese og lipid dråpe (ld) dannelse og fusjon. Caveolin – 1 Og Cavin-1 er nødvendige for dannelse og stabilisering av caveolae. Mutasjon I CAV1 (TYPE 3) ELLER CAVIN1 (type 4) kan føre til tap av caveolae i membranen. Nc, nucleus. Endoplasmatisk retikulum. Mc, mitokondrier. * Lipin er et cytosolisk enzym forankret av seipin i ER.
disse reaksjonene forekommer i adipocyttenes endoplasmatiske retikulum (ER), hvor en progressiv akkumulering av triglyserider fører til dannelse av små lipiddråper (LD)8. Produktet av genet BSCL2 er et transmembranprotein kalt seipin som forårsaker fusjon av liten LD, som stammer fra stor LD. Seipin ligger i ER og konsentrerer seg ved krysset MED nascent LD, noe som letter lipidtrafikken mellom ER og LD og inkorporeringen av triglyserider I LD9. Seipin kan også fungere som et er-anker til det cytosoliske enzymet lipin 1. Foruten å være nødvendig for lipid dråpe fusjon, størrelse og morfologi, er seipin også viktig for adipogenese (via interaksjon til lipin 1) og cellulære triglyserid lipolysis10, 11. Mangel på seipin hindrer differensiering av pre-adipocytter til adipocytter og påvirker den endelige modningen9, som vist ved studier i mesenkymale stamceller med bscl2 slått ut12. Ikke-fettvev uttrykker også seipin, og andre funksjoner skal bestemmes.
i adipocytter, caveolae, som er spesialiserte 50-100nm membran invaginations, står for 20% av plasmamembranen området, noe som gjør adipocytter cellene med høyest tetthet av caveolae13. Dannelsen av lipiddråper trenger et membranprotein ( Caveolin-hovedkomponenten av caveolae membraner) og et cytoplasmatisk protein (Cavin-1)14. Generene CAV1, CAV2 og CAV3 koder for tre former for caveolin med lignende strukturer (Henholdsvis Caveolin-1, Caveolin-2 og Caveolin-3). Caveolin-1 Og Caveolin-2 finnes i adipocytter, fibroblast og endotelceller, Og Caveolin-3 finnes bare i skjelett-og hjertemuskel13, 15. Caveolin-1 er den viktigste og mest studerte. Det uttrykkes i to forskjellige isoformer (1a og 1b). Caveolin-1 translokerer fra plasmamembranen til lipiddråpe, som er nødvendig for lipidhandel og metabolisme16. Lipiddråper lagrer triglyserider etter mating, og disse molekylene hydrolyseres til fettsyre og frigjøres under fasting; denne mekanismen kan reguleres av Caveolin-116. Caveolin-1-mangel øker også følsomheten for celledød ved autofagi17.
GENET CAVIN1 koder for et cytoplasmatisk protein kalt caveolae assosiert protein 1 (Cavin-1) 14, 16, som er obligatorisk for dannelse og stabilisering av caveolae. Cavin-1 uttrykkes i adipocytter, muskelceller og andre celler, og er også viktig i overføringen av caveolae-opprinnelige signaler14, 18. Knockout AV CAV1-genet forårsaker mangel på caveolae i ikke-muskelceller, mens knockout AV CAVIN1 forårsaker fravær av caveolae i alle vev, inkludert muskel14. Mangelen på caveolae kan påvirke regulering av lipolyse, fettsyreflux, triglyseridsyntese og signaler fra andre veier.
Typer CGL
basert på påviselige genetiske endringer, er fire typer beskrevet. Type 1 og 2 er ansvarlig for over 95% av tilfellene, og type 2 har en mer alvorlig påvirket fenotype. Bare ett tilfelle av type 3 og rundt 30 tilfeller av type 4 har blitt rapportert4.
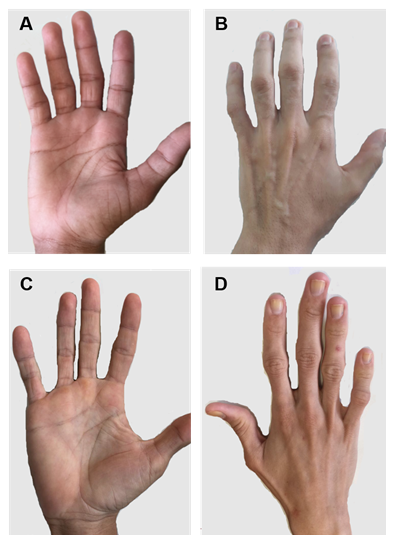
Figur 2. Pasienter med CGL type 1 og 2. (A) Og (B) anterior og posterior utsikt over hender av type 1 pasienter. Tilsynelatende normale hender, siden det fortsatt er mekanisk fettvev. (C) Og (D) Anterior og posterior utsikt over hender av type 2 pasienter. Alvorlighetsgraden av sykdommen er større, og mangel på fett er tydelig og lett merkbar.
CGL Type 1. I 1999, Garg et al. beskrevet pasientenes mutasjon på kromosom 9q34, og tre år senere Agarwal et al. viste AGPAT2 som enzymet påvirket av denne mutasjonen2, 19. På grunn AV mutasjon AV DENNE AGPAT2, ingen eller minimal produksjon av triacylglycerol skjer ved stimulans av andre isoformer. Fenotypen AV AGPAT2 knockout-mus er lik den hos mennesker MED CGL-type, som bekrefter dette enzymets rolle i patofysiologien20, 21.
CGL Type 2. Magre et al. var de første til å identifisere mutasjonen i seipin-genet (kromosom 11q13) 3. Mutasjoner (for det meste tull) av seipin-genet (BSCL2) produserer et avkortet protein og kan påvirke lipidmetabolismen ved forskjellige mekanismer: a) reduksjon i seipinstabilitet; b) reduksjon i evnen til å binde lipin 1; og c) manglende oligomerisering og lokalisere seg utelukkende til er-membranen11. Noen celler er fortsatt i stand til å generere triacylglycerol og små lipiddråper, men store lipiddråper er fraværende på grunn av tap av evnen til fusjon av disse små lipiddråper. Det er også en svikt i ekspresjonen av adipogene faktorer, som peroksisomproliferatoraktivert reseptor gamma (PPARG), samt adiponektin og adipocytfettsyrebindende protein (FABP4) 11, 16. Seipin mangel svekker adipogenese, øker lipolyse og forhindrer triglyserider akkumulering i adipocytter.
CGL type 3. Denne typen ble nylig beskrevet hos en pasient som til tross FOR Å ha CGL-fenotype ikke hadde mutasjoner I gener AGPAT2 ELLER BSCL222. Mus med En Mutasjon I Cav1 er resistente mot diettindusert fedme og har insulinresistens, hypertriglyseridemi, redusert adiponektin, redusert fettmasse og små adipocytter16. Etter å ha valgt kandidatgener basert på studier på mus, Kim Et al. bekreftet tilstedeværelsen av en nonsensmutasjon i caveolin – 1-genet (CAV1), på kromosom 7q3122.
CGL type 4. I dette er en sjelden type det berørte genet ER CAVIN1, som koder For Proteinet Cavin-1. Hos mennesker er det rapportert hos pasienter med generalisert medfødt lipodystrofi og muskeldystrofi15, 23.
nylig har mutasjoner I PCYT1A-og pparg-genene også blitt beskrevet som forårsaker lipodystrofi24, 25.
Kliniske trekk
CGL-pasienter presenterer vanligvis akromegaloide facies, acanthosis nigricans, phebomegali, hepatomegali og muskulær hypertrofi5, 26, 27. Flere forfattere citerer navlestreng som et klinisk funn av syndromet26. Vi evaluerte hyppigheten av det i vår serie av pasienter, og ingen av dem presenterte denne endringen28. Faktisk forårsaker fraværet av periumbilisk fettvev fremspring av navlestrengen, og dette kan feilaktig diagnostiseres som en brokk28, 29.
når adipocytter ikke kan lagre fett tilstrekkelig, akkumuleres det i andre vev som lever og muskler, noe som forårsaker alvorlig insulinresistens. Bentetthetometri (DXA) kan vise normal eller høy benmineraltetthet30 og redusert total kroppsfett (vanligvis lavere enn 6%)27. Som følge av lavt kroppsfett er serum adiponektin og leptin lavt også27. Som leptin er viktig for å kontrollere sult, har disse pasientene vanligvis hyperfagi, som er lett synlig siden barndommen. Adiponektin spiller en viktig rolle som insulinsensibilisator, og mangelen forverrer insulinresistensen. Til tross for dette er glukose og glykert hemoglobin i utgangspunktet normalt på bekostning av svært høye insulinnivåer. Diabetes starter vanligvis i puberteten; i vår serie var gjennomsnittsalderen for debut 15,8±7,1 år27. I utgangspunktet kontrolleres de med orale legemidler, som trenger høye doser insulin om noen år27. Arteriell hypertensjon forekommer hos en tredjedel av pasientene27.
det er noen spesifikke kliniske trekk ved HVER CGL-type. Pasienter med type 1 fortsatt tilstede mekanisk fettfett, spesielt i palmer, såler, orbital, peri-artikulære regioner31. I motsetning viser type 2-pasienter et fravær av metabolsk og mekanisk fettvev. Seipin er sterkt uttrykt i hjernen og cerebellum og er også involvert i regulering av nevrale funksjoner. Mer enn halvparten av type 2-pasientene har noe kognitiv funksjonsnedsettelse1, 8. Type 3 og 4 har bevaring av mekanisk og benmargsfett, og type 4 har muskelsvakhet forbundet med høy serum kreatinkinase og spinal ustabilitet15.
det er også kjønnsspesifikke kliniske trekk. Polycystiske ovarier og amenorrhea er vanlige32. Menstruasjonssykluser vanligvis tilbake til det normale ved bruk av metreleptin, sannsynligvis på grunn av forbedring i insulinfølsomhet og restaurering AV lh pulsatilitet32. Type 2 menn kan ha teratozoospermi på grunn av mangel på seipin i bakterieceller33.
Hypertriglyseridemi oppstår siden de første årene av livet og kan forårsake akutt pankreatitt. HDL er vanligvis lavere enn 30 mg / dL. Forhøyninger av leverenzymer er også et tidlig funn og kommer fra fettavsetningen i leveren. Progressiv reduksjon i serumplatetall tyder på forverring av leversykdom og sannsynlig cirrhose34.
Siden Cavin-1 finnes i muskelcellene, har pasienter med type 4 mild muskelsvakhet og forhøyet kreatinkinase15.
Forventet Levealder, hovedsakelig i type 2, er betydelig redusert, med dødsfall som ikke sjelden forekommer før fylte 30 år (personlig erfaring basert på 20 pasienter som døde de siste 19 årene). Dødsårsakene er relatert til diabetes (nyresvikt, plutselig død), lever (skrumplever, fordøyelsesblødning) eller infeksjoner.
Diagnose Og Behandling
CGL-diagnosen er basert på kliniske data: akromegaloide egenskaper, acanthosis nigricans, reduksjon av totalt kroppsfett, muskelhypertrofi og fremspring av navlestrengen. Laboratoriedata kan også vise diabetes med alvorlig insulinresistens og hypertriglyseridemi. Imaging tester kan bidra til å identifisere ektopiske forekomster av fett hovedsakelig i leveren og bukspyttkjertelen(hepatisk steatose med hepatomegali og pankreatisk steatose). DXA kan bekrefte lavt kroppsfett og høy bentetthet30.
behandlingen av CGL består av streng kontroll av dietten med reduksjon av inntaket av fett, hovedsakelig triglyserider og matvarer med høy glykemisk indeks for å forebygge og kontrollere komorbiditeter29. Imidlertid er det ideelle dietten et utfordrende mål å oppnå på grunn av økt appetitt og den alvorlige begrensningen som er anbefalt. Fysisk aktivitet bør også oppmuntres til å forbedre kontrollen av komorbiditet, unntatt hos pasienter med kontraindikasjoner som alvorlig kardiomyopati29.
Når det Gjelder narkotikabehandling, kan disse pasientene behandles med de vanlige medisinene for diabetes, hypertensjon og dyslipidemi retningslinjer. Førstevalget for behandling av diabetes og insulinresistens er metformin, men vanligvis er det ikke nok. I motsetning til behandling av delvis lipodystrofi, bør tiazolidindioner brukes med forsiktighet29. Andre orale antidiabetika brukes, men de ble ikke spesifikt studert hos CGL-pasienter. Det er data fra dyr som tyder på at BRUK AV sglt2-hemmere (dapagliflozin) kan ha fordeler som forhindrer kardiomyopati35; studier er nødvendig for å bekrefte dette hos mennesker. Når sykdommen utvikler seg og alvorlig insulinresistens oppstår, er det nødvendig med høye daglige doser insulin. Mangelen på subkutan fettvev er et problem ved administrering av høye doser insulin. Mer konsentrert insulin (U-300 Eller U500) kan være nødvendig36. Disse pasientene har alvorlig dyslipidemi, hovedsakelig på grunn av økning av triglyserider og lav HDL, og derfor er bruk av fibrat noen ganger nødvendig for å forhindre akutt pankreatitt. I tillegg, på grunn av den høye kardiovaskulære risikoen hos disse pasientene, bør intervensjon med et statin vurderes, OG MÅLENE FOR LDL eller ikke-HDL bør være strenge29.
Daglige injeksjoner av metreleptin forårsaker en betydelig reduksjon i appetitten og gir fordeler ved å senke glykemi, triglyseridemi og leverenzymer. Det er bemerkelsesverdig, spesielt hos barn, reduksjon av abdominal omkrets, sannsynligvis på grunn av reduksjon av hepatomegali.
Konklusjon
CGL er en sjelden og alvorlig sykdom som kan oppstå med diabetes (vanligvis krever høye doser insulin) og tidlig død. Fenotypen til pasienten er ganske karakteristisk, og krever imidlertid kunnskap om syndromet av helsepersonell for å gjøre en tidlig diagnose. Metreleptin synes å være den eneste medisinen for øyeblikket som kan endre sykdommens naturlige historie.
Interessekonflikt: ingen.
- Nolis T. Utforske patofysiologien bak de mer vanlige genetiske og oppkjøpte lipodystrofiene. Tidsskrift for den norske legeforening. 2014 Jan; 59(1): 16-23.
- Agarwal AK, Arioglu E, De Almeida S, et al. AGPAT2 er mutert ved medfødt generalisert lipodystrofi knyttet til kromosom 9q34. Nat Genet. 2002 Mai; 31(1): 21-3.
- Magre J, Delepine M, Khallouf E, Et al. Identifikasjon av genet endret I berardinelli-Seip medfødt lipodystrofi på kromosom 11q13. Naturgenetikk. 2001 August; 28 (4): 365-70.
- Patni N, Garg A. Medfødte generaliserte lipodystrofier-ny innsikt i metabolsk dysfunksjon. Natur vurderinger Endokrinologi. 2015 September; 11 (9): 522-34.
- Garg A. Ervervet og arvet lipodystrofier. New England journal of medicine (engelsk). 2004 Mar 18; 350 (12): 1220-34.
- De Azevedo Medeiros LB, Candido Dantas VK, Craveiro Sarmento AS, Et al. Høy forekomst Av Berardinelli-Seip Medfødt Lipodystrofi I Rio Grande do Norte-Staten, Nordøst-Brasil. Diabetol Metab Syndr. 2017; 9: 80.
- Chiquette E, Muntlig EA, Garg A, et al. Estimering av forekomsten av generalisert og delvis lipodystrofi: funn og utfordringer. Diabetes, Metabolsk Syndrom Og Fedme: Mål Og Terapi. 2017: 375-83.
- Wee K, Yang W, Sugii S, Et al. Mot en mekanistisk forståelse av lipodystrofi og seipin-funksjoner. Biovitenskap rapporter. 2014; 34(5).
- Dukken L, Magre J, Cariou B, et al. Funksjon av seipin: ny innsikt Fra Bscl2 / seipin knockout musemodeller. Biochimie. 2014 Jan; 96: 166-72.
- Sim MF, Dennis RJ, Aubry EM, Et al. Det humane lipodystrofieproteinet seipin er en er-membranadapter for adipogen PA fosfatase lipin 1. Molekylær metabolisme. 2012; 2(1): 38-46.
- Sim MF, Talukder MM, Dennis RJ, Et al. Analyse av naturlig forekommende mutasjoner i humant lipodystrofi protein seipin avslører flere potensielle patogene mekanismer. Diabetologia. 2013 November; 56 (11): 2498-506.
- Payne VA, Grimsey N, Tuthill A, et al. Det humane lipodystrofigenet BSCL2 / seipin kan være avgjørende for normal adipocyttdifferensiering. Diabetes. 2008 August; 57 (8): 2055-60.
- Cohen AW, Hnasko R, Schubert W, et al. Rolle caveolae og caveolins i helse og sykdom. Fysiologiske vurderinger. 2004 Oktober; 84 (4): 1341-79.
- Pilch PF, Liu L. Fett grotter: caveolae, lipid handel og lipid metabolisme i adipocytter. Trender i endokrinologi og metabolisme: TEM. 2011 August; 22 (8): 318-24.
- Hayashi YK, Matsuda C, Ogawa M, et al. Humane ptrf-mutasjoner forårsaker sekundær mangel på caveoliner som resulterer i muskeldystrofi med generalisert lipodystrofi. J Clin Invest. 2009 September; 119 (9): 2623-33.
- Parton RG, del Pozo MA. Caveolae som plasmamembran sensorer, beskyttere og arrangører. Natur vurderinger Molekylær cellebiologi. 2013 Februar; 14 (2): 98-112.
- Legg S, Briand N, Blouin CM, et al. Den lipoatrofiske caveolin – 1-mangelfulle musemodellen avslører autofagi i modne adipocytter. Autofagi. 2010 August; 6 (6): 754-63.
- Liu L, Brown D, McKee M, et al. Sletting Av Cavin / PTRF forårsaker globalt tap av caveolae, dyslipidemi og glukoseintoleranse. Cellemetabolisme. 2008 Oktober; 8 (4): 310-7.
- Garg A, Wilson R, Barnes R, et al. Et gen for medfødt generalisert lipodystrofi kart til menneskelig kromosom 9q34. Tidsskriftet Journal of clinical endocrinology and metabolism. 1999 September; 84 (9): 3390-4.
- Vogel P, Les R, Hansen G, Et al. Patologi av medfødt generalisert lipodystrofi I Agpat2 – / – mus. Veterinærpatologi. 2011 Mai; 48 (3): 642-54.
- Cortes VA, Curtis DE, Sukumaran S, et al. Molekylære mekanismer for hepatisk steatose og insulinresistens I AGPAT2-mangelfull musemodell av medfødt generalisert lipodystrofi. Cellemetabolisme. 2009 Februar; 9 (2): 165-76.
- Kim CA, Delepine M, Boutet E, Et al. Forening av en homozygot nonsens caveolin – 1 mutasjon Med Berardinelli-Seip medfødt lipodystrofi. J Clin Endocrinol Metab. 2008 April; 93 (4): 1129-34.
- Rajab A, Straub V, McCann LJ, Et al. Fatal hjertearytmi og lang QT-syndrom i en ny form for medfødt generalisert lipodystrofi med muskelkrypling (CGL4) på GRUNN AV ptrf-CAVIN mutasjoner. PLoS genetikk. 2010 Mar 12; 6 (3): e1000874.
- Payne F, Lim K, Girousse A, et al. Mutasjoner som forstyrrer Kennedy fosfatidylkolinveien hos mennesker med medfødt lipodystrofi og fettleversykdom. Proc Natl Acad Sci Usa 2014 Juni 17; 111 (24): 8901-6.
- Dyment DA, Gibson WT, Huang L, et al. Bialleliske mutasjoner ved PPARG forårsaker en medfødt, generalisert lipodystrofi som Ligner berardinelli-Seip syndromet. Eur J Med Genet. 2014 September; 57 (9): 524-6.
- Garg A. Klinisk gjennomgang#: Lipodystrofier: genetiske og oppkjøpte kroppsfettforstyrrelser. Tidsskriftet Journal of clinical endocrinology and metabolism. 2011 November; 96 (11): 3313-25.
- Lima JG, Nobrega LH, De Lima NN, Et al. Kliniske og laboratoriedata fra en stor serie pasienter med medfødt generalisert lipodystrofi. Diabetol Metab Syndr. 2016; 8: 23.
- Lima GJ, Lima NN, Oliveira CF, Et al. Umbilical Brokk Hos Pasienter Med Berardinelliseip Syndrom: Er Det Virkelig Brokk. J Clin Mol Endocrinol. 2015; 1(1): 3.
- Brun RJ, Araujo-Vilar D, Cheung PT, Et al. Diagnostisering Og Behandling Av Lipodystrofi Syndromer: En Multi-Samfunnet Praksis Retningslinje. J Clin Endocrinol Metab. 2016 Desember; 101( 12): 4500-11.
- Lima JG, Nobrega LH, Lima NN, Et al. Bentetthet Hos Pasienter Med Berardinelli-Seip Medfødt Lipodystrofi Er Høyere I Trabekulære Steder og Hos Type 2 Pasienter. J Clin Densitom. 2016 November 25.
- Simha V, Garg A. Fenotypisk heterogenitet i kroppsfettfordeling hos pasienter med medfødt generalisert lipodystrofi forårsaket av mutasjoner I agpat2-eller seipin-gener. J Clin Endocrinol Metab. 2003 November; 88 (11): 5433-7.
- Musso C, Cochran E, Javor E, Et al. Den langsiktige effekten av rekombinant metionyl human leptinbehandling på hyperandrogenisme og menstruasjonsfunksjon hos hunn-og hypofysefunksjon hos mannlige og kvinnelige hypoleptinemiske lipodystrofiske pasienter. Stoffskifte. 2005 Februar; 54 (2): 255-63.
- Jiang M, Gao M, Wu C, et al. Mangel på testikulær seipin forårsaker teratozoospermi syndrom hos menn. Proc Natl Acad Sci Usa 2014 Mai 13; 111 (19): 7054-9.
- Mitchell O, Feldman DM, Diakow M, et al. Patofysiologien av trombocytopeni i kronisk leversykdom. Hepat Med. 2016; 8: 39-50.
- Joubert M, Jagu B, Montaigne D, et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes. 2017 Apr; 66(4): 1030-40.
- Lima JG, Lima NN, Lima RLM, et al. Glargine U300 Insulin as a Better Option than Degludec U100 to Treat a Congenital Generalized Lipodystrophy Patient. Clin Diabetes Res. 2017; 1(1).