Autosomal Recessiv Medfødt Iktyose / Actas Dermo-Sifiliográ
Innledning
den siste konsensusklassifiseringen av iktyose skiller mellom 2 hovedformer: de ikke-syndromiske formene, som kun presenterer hudmanifestasjoner, og syndromformene, som også presenterer manifestasjoner i andre organer (Tabell 1).1 blant ikke-syndromiske former er 4 grupper identifisert: vanlige ichthyoser, autosomale recessive medfødte Ichthyoser (ARCIs), keratinopatiske ichthyoser og andre mindre vanlige ichthyoser.Tradisjonelt Ble Gruppen Av ARCIs delt inn i 2 lidelser, lamellær ichthyosis (LI) og medfødt ichthyosiform erytroderma (CIE). I den nye klassifiseringen ble harlekin iktyose (HI) lagt til denne gruppen1 fordi inaktiverende mutasjoner I abca12-genet har blitt identifisert som ansvarlig for denne lidelsen,2,3 mens nonsensmutasjoner i samme gen kan gi OPPHAV TIL li4 eller CIE5,6 fenotype. Andre mindre vanlige varianter som inngår I Gruppen ARCIs er selvhelbredende collodion baby (SHCB), acral SHCB og badedrakt ichthyosis.7-9
Konsensusklassifisering Basert På De Kliniske Egenskapene Til Ichthyosis1.
| Nonddromiske Former | Syndromiske Former |
| Felles ichthyosesichthyosis vulgarisrecessiv x-bundet ichthyosis (nonsyndromic )ajimajor formsharlekin ichthyosisLamellar ichthyosismedfødt ichthyosiform erytrodermaminor formerselvhelbredende collodion babyAcral selvhelbredende collodion babybading dress ichthyosisKeratinopathic Ichthyosestor formsEpidermolytic Ichthyosisoverfladisk epidermolytic ichthyosisminor formsannulær epidermolytic ichthyosecurth-Macklin Ichthyosisautosomal recessiv epidermolytisk ichthyosisepidermolytisk nevusandre Formerlorikrin keratodermaerytrokeratodermi vararabilispeeling hudsyndrommedfødt retikulær ichthyosiform erytrodermaklick syndrom | Syndromisk X-bundet Ichthyosrecessiv x-bundet ichthyosrecessiv x-bundet ichthyosis (syndromisk)ichthyosis follicularis, alopecia og fotofobi (IFAP) syndromkonradi-Scammann-happle syndrom (chondrodysplasia punctata type 2)syndromisk autosomal iktyosehudforstyrrelsernetonsyndromichthyosis-hypotrichosis syndromichthyosis-skleroserende kolangitt syndromtrichothystrophyneurologiske lidelsersjö-larsson syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, mental retardasjon, enteropati, døvhet, perifer nevropati, ichthyosis, keratoderma.
kun begrensede data er tilgjengelige på epidemiologi Av ARCIs. I Usa er det estimert en prevalens ved fødselen på 1 per 100 000 innbyggere FOR LI og 1 per 200 000 innbyggere for CIE. Andre studier har rapportert en kombinert prevalens FOR LI og CIE på 1 per 200 000 til 300 000 populasjoner.10,11 i enkelte land som Norge er estimert prevalens større (1 per 91 000) på grunn av grunnleggermutasjoner.12 funnet av 1 eller flere tilbakevendende mutasjoner i en populasjon kan skyldes at mutasjonen skjedde på et gitt tidspunkt i historien og deretter ble overført fra generasjon til generasjon (grunnleggermutasjon) eller fordi regionen av genomet der mutasjonen er funnet, har EN DNA-sekvens som er mottakelig for mutasjon (mutation hotspot). I Spania er den estimerte prevalensen AV ARCI 1 per 138 000 i den generelle befolkningen og 1 per 61 700 blant barn under 10 år.13 i enkelte Regioner I Spania kan forekomsten være enda høyere. På den Galiciske kysten ble for eksempel en prevalens på 1 per 33 000 rapportert, også på grunn av en grunnleggereffekt.14
Lamellær Iktyose Og Medfødte Iktyosiform Erytrodermakliniske Egenskaper
Selv om DET opprinnelig ble antatt AT LI og CIE var forskjellige enheter, har det vært rapporter om pasienter med mellomliggende kliniske manifestasjoner, og begge tilstandene kan skyldes mutasjoner i samme gen.15,16 i tillegg kan pasienter med samme mutasjon, selv innenfor samme familie, utvikle forskjellige fenotyper.12,15
de fleste pasienter er født innhyllet i en kollodionmembran som gradvis forsvinner i løpet av de første ukene av livet og erstattes av den endelige fenotypen (Fig. 1A). Hypohidrose, alvorlig varmeintoleranse og negledystrofi observeres ofte i BÅDE LI og CIE.17-19 Pasienter med LI har vanligvis mer alvorlige kliniske manifestasjoner enn DE med CIE. De har store platelike skalaer, ofte av mørk farge, som dekker hele kroppsoverflaten. Erythroderma er enten fraværende eller minimal. Slike pasienter har vanligvis ektropion og, til tider, eclabium, hypoplasi av ledd og nesebrusk, arrdannelse alopecia, spesielt ved kanten av hodebunnen, og palmoplantar keratoderma (Fig. 1B Og C). CIE er preget av tilstedeværelsen av erythroderma og fin hvitaktig skalering (Fig. 2). Noen pasienter har markert erytem og generalisert skalering. Vektene kan være store og mørke, spesielt på ekstensorflatene på beina. I mindre alvorlige tilfeller er erytem mild og skaleringen er fin.

Kliniske egenskaper av lamellær iktyose. A, Brunaktig lamellar desquamation. B, Merket plantar hyperkeratose. C, Arrdannelse alopecia i hodebunnen.
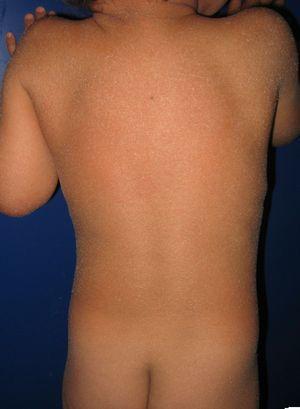
Pasient med medfødt ichthyosiform erytroderma og mutasjoner I aloxe3-genet. Mild erytem og generalisert hvitaktig furfuraceous desquamation kan ses.
Histopatologi
Histopatologiske endringer gir ingen diagnose. I LI observeres massiv orthokeratotisk hyperkeratose, vanligvis med to ganger forlengelsen som I CIE. Epidermis er acanthotic og noen ganger tar på seg en psoriasis-lignende utseende. Celleproliferasjonshastigheten er normal eller litt forhøyet.17-19 Pasienter MED CIE har mindre markert hyperkeratose, med fokal eller omfattende parakeratose, et normalt eller fortykket granulært lag og mer uttalt akanthose. Den epidermale omsetningen økes.17-19
Ultrastructure
selv om en nær sammenheng mellom molekylære, kliniske og ultrastructural funn hittil ikke er funnet, kan elektronmikroskopi likevel være nyttig for å utelukke andre former for iktyose og for å lede genetiske analyser i noen tilfeller. Fire typer medfødt ichthyosis er beskrevet (Tabell 2).
Ultrastrukturell Klassifisering Av Medfødte Ichthyoses.
| Type | Hovedfunksjon | Andre Funksjoner | Mutasjoner | Kliniske Manifestasjoner |
| 1 | Fravær av ultrastrukturelle markører av iktyose type 2, 3 og 4 | Lipiddråper eller ringer i stratum corneum (hyppigst)små keratohyalin granulesVesicular eller lobular membran belegg granulater | TGM1 (33.3%)ALOX12B (2 tilfeller) | CIE |
| 2 | Kolesterol spalter i stratum corneum | Fravær Eller tynning av cornified konvoluttsmå keratohyalin granulesLipid dråper | TGM1 (89-100%) | LI |
| 3 | Laminerte membranstrukturer i stratum granulosum og / eller stratum corneum. | Unormale membranbelegg granuleslipiddråperfoci av fremtredende juxtanukleære vakuoler i det granulære laget | NIPAL4 (93%) | CIE (hyppigst)LI |
| 4 | Trilamellære membranpakker som fyller noen celler i stratum granulosum og / eller stratum corneum | Unormale membranbelegg granulater | FTAP4 | Iktyose prematuritetssyndrom(100%) |
Forkortelser: CIE, medfødt ichthyosiform erytroderma; LI, lamellær ichthyosis.
Medfødt Iktyose Type 1
Medfødt iktyose type 1 er karakterisert ved fravær av ultrastrukturelle markører for iktyose type 2, 3 og 4. Derfor blir diagnosen vanligvis bare gjort når de andre typene er utelukket. Det hyppigste funnet er tilstedeværelsen av lipiddråper eller ringer i stratum corneum (Fig. 3A).20 disse lipiddråpene er ikke en konstant funksjon eller spesifikk for denne typen, da de ikke er tilstede i alle tilfeller,20 og de kan være tilstede i andre typer ichthyosis.21,22 Klinisk presenterer de fleste pasienter med manifestasjoner AV CIE.12,20 en tredjedel av pasientene har mutasjoner I tgm1-genet.16 denne ultrastrukturelle typen har også blitt identifisert i forbindelse med mutasjoner I alox12b-genet.23,24

Elektronmikroskopbilder. A, Medfødt iktyose type 1, som viser lipiddråper i stratum corneum og fravær av ultrastrukturelle markører av de andre typer iktyose. B, Medfødt ichthyosis type 2, preget av tilstedeværelsen av kolesterolklyver (pil) i corneocytter.
Medfødt Iktyose Type 2
Medfødt iktyose type 2 er karakterisert ved kolesterol spalter i stratum corneum (Fig. 3B).21 Slike kløfter er et konstant funn i denne typen iktyose, og kan påvises i forskjellige biopsier hos samme pasient; behandling med orale retinoider har ingen innvirkning på disse kløftene.12,25 elektrontette aggregater er også observert på korneocytter hos noen pasienter med mangelfull TGase 1-aktivitet.26-28 Klinisk presenterer de fleste pasienter med alvorlige manifestasjoner AV CIE.12 denne ultrastrukturelle typen er sterkt assosiert med mutasjoner I tgm1-genet.12,16
Medfødt Iktyose Type 3
Medfødt iktyose type 3 er preget av lamellære membranøse strukturer i stratum granulosum og / eller stratum corneum. Disse strukturene er arrangert i strimler rundt et tomt rom nær kjernen.22,29-31 de kliniske manifestasjonene i denne typen er forskjellige fra de andre; utbruddet av iktyose er variabel, desquamation og erytem kan være ujevn eller generalisert, og spesielt bøyningene påvirkes. Mutasjoner I nipal4-genet er ansvarlige for 93% av ichthyoses type 3.32
Medfødt Iktyose Type 4
Karakteristisk, i medfødt iktyose type 4, er noen celler i stratum granulosum og stratum corneum fylt med trilamellære membranpakker.33 disse funnene er patognomisk for ichthyosis prematurity syndrome, en tilstand som for tiden anses som en syndromisk form for ichthyosis.34,35
Molekylære Studier
i genetiske termer Er ARCIs svært heterogene. TGM1-genet er assosiert med de fleste tilfeller, men mutasjoner i 5 andre gener (ALOX12B, ALOXE3, NIPAL4, CYP4F22 OG ABCA12) er rapportert. Fischer et al.36 studerte 520 familier MED ARCI og identifiserte mutasjoner i minst 1 av disse genene i 78% av tilfellene (TGM1 hos 32%, NIPAL4 hos 16%, ALOX12B hos 12%, CYP4F22 hos 8%, ALOXE3 hos 5% OG ABCA12 hos 5%). I en annen studie med 250 pasienter MED ARCI av forskjellig opprinnelse hadde 38% tgm1-mutasjoner, 6,8% hadde aloxe3-mutasjoner og 6,8% hadde alox12b-mutasjoner.37 I Galicia identifiserte vi mutasjoner I GENENE TGM1, ALOX12B, ALOXE3, NIPAL4 og CYP4F22 i 75% av familiene som ble studert, men fordelingen av mutasjoner var forskjellig.14 TGM1-genet ble mutert i 68.7% av tilfellene MENS aloxe3-genet ble mutert i bare 1 pasient. Vi oppdaget ikke mutasjoner i noen av de andre 3 genene som ble studert.
TGM1
tgm1-genet ligger på kromosom 14q11. 2 og har 15 eksoner (GenBank NM-000359.2). Det koder For TGase 1-enzymet, som er et av de 3 tgase-enzymene som finnes i epidermis.38 dette enzymet deltar i dannelsen av den cornified konvolutten ved å katalysere kalsiumavhengig kryssbinding av flere proteiner som involucrin, loricrin og prolinrike proteiner.39,40 katalyserer det ogsa bindingen av ??- hydroksyceramider i det ytre laget av den cornified konvolutten med proteiner i det indre laget.41,42 hos pasienter MED tgm1-mutasjoner mangler den kornede konvolutten og TGase 1-aktiviteten er redusert eller ikke-eksisterende.43-47
siden 1995,da dette genet ble identifisert som ansvarlig for NOEN TILFELLER AV ARCI, har 48-50 mer enn 110 mutasjoner blitt rapportert hos pasienter av forskjellig opprinnelse. Mutasjoner I TGM1 er den vanligste årsaken TIL ARCI.36,37 denne mutasjonen er funnet i 55% av tilfellene I Usa og i 84% av tilfellene I Norge.12,51 den hyppigste mutasjonen er c.877-2A> G, som er funnet i 34% av de muterte alleler rapportert til dags dato.52 den høye frekvensen av denne mutasjonen i land som Usa og Norge skyldes en grunnleggereffekt.12,53 den nest hyppigste mutasjonen er P.Arg142His. Denne og lignende mutasjoner har blitt rapportert i Land som Egypt, Tyskland, Finland og Usa, 15,49-51, 54-56, og det ser ut til at disse er hotspot mutasjoner.57 p. Arg307Trp-mutasjonen er hyppig i Den Japanske befolkningen.5 I Galicia, s. Arg760X, ca. 1223_1227delACACA og ca.984 + 1g>a mutasjoner I TGM1 ble identifisert i 81,82% av familiene med mutasjoner i dette genet, noe som tyder på en grunnleggereffekt.14 Bekreftelse av denne hypotesen ble oppnådd ved haplotype studie (arbeid som ennå ikke publisert).
tgm1 mutasjoner er ansvarlige for DE fleste tilfeller AV LI15,27,44,46,56,58-63 og for en liten prosentandel av TILFELLENE AV CIE.43,47,64,65 slike mutasjoner kan også gi opphav til ANDRE former FOR ARCI som SHCB, acral SHCB, og badedrakt iktyose.
Mange studier har forsøkt å demonstrere genotype-fenotypeforbindelser mellom mutasjoner I TGM1 og ultrastrukturelle eller kliniske funn, men ingen signifikant korrelasjon er observert til dags dato.15,16,53 generelt er pasienter med mutasjoner I tgm1-genet mer alvorlig påvirket enn de uten slike mutasjoner. I en studie med 83 PASIENTER MED ARCI I Sverige og Estland var forekomsten av ektropion og collodion baby assosiert MED tgm1-mutasjoner, mens en høyere forekomst av erytem ble observert hos pasienter uten mutasjoner i dette genet.66 En annen studie viste at typen skalering er hovedforskjellen mellom bærere og ikke-bærere AV tgm1-mutasjoner, ved å finne at alle pasienter med mutasjoner i dette genet hadde lamellær skalering mens 80% av de uten TGM1-mutasjoner hadde fin skalering.14 i tillegg har det blitt sett at trunkerende mutasjoner oftere er assosiert med hypohidrose og svetteforstyrrelser enn missense mutasjoner.51 i den nordamerikanske befolkningen forutsier en modell basert på tilstedeværelsen av visse kliniske egenskaper at pasienter som er født som kollodionbarn og har okulære lidelser og / eller alopecia, er 4 ganger mer sannsynlig Å ha tgm1-mutasjoner.51
aloxe3 og ALOX12B
aloxe3 og alox12b gener er plassert på kromosom 17p13. 1. 67 de har en lignende struktur med 15 eksoner som koder for epidermal LOXs eLOX-3 og 12r-LOX.68,69 det faktum at de hovedsakelig uttrykkes i de suprabasale lagene i epidermis, støtter deres rolle i avanserte faser av epidermal differensiering, med deltakelse i behandlingen av lamellære legemer.24,70 disse enzymene virker på tilstøtende trinn i hepoksilinbanen(Fig. 4). 12R-LOX omdanner arakidonsyre TIL 12r-hydroksyeicosatetraensyre mens eLOX-3 omdanner dette produktet til en epoksyalkoholisomer 69, 71 i hepoxilin A3-familien.72 hepoksilinproduktet er ustabilt og hydrolyseres i celler til et spesifikt trihydroksyderivat (trioksilin). Selv om den nøyaktige rollen til produktene i hepoksilinbanen ikke er kjent, har det vært spekulert i at de kan delta i dannelsen av intercellulære lipider i stratum corneum eller fungere som signaler for å indusere keratinocytdifferensiering.
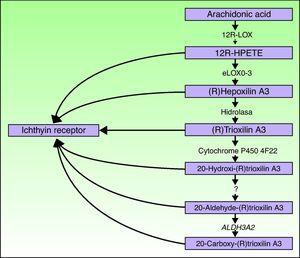
Skjematisk diagram av hepoksilinbanen, som viser deltakelsen AV aloxe3 -, ALOX12B -, NIPAL4-og CYP4F22-genene. Mutasjoner i disse genene er ansvarlige for noen TYPER ARCI. HPETE indikerer hydroperoksyikosatetraensyre.
alox12b-og ALOXE3-genene ble først identifisert i 2002.73, 74 Siden da har mer enn 30 mutasjoner I alox12b-genet23,24,37,75-77 og omtrent 10 I ALOXE3-genet37,74,75 blitt rapportert. Disse mutasjonene er ansvarlige for 14% til 17% Av ARCIs36,37 og 72.2% Av SHCBs.23,78,79 det kausative forholdet mellom disse mutasjonene og fenotypen ble bekreftet ved å demonstrere at katalytisk aktivitet av epidermal LOX ble fullstendig avskaffet hos pasienter med disse mutasjonene75,80 og ved bruk av dyremodeller som reproduserte ichthyosiform fenotype sett hos mennesker.81-83 begge gener er ansvarlige for en tilsvarende prosentandel AV ARCI-tilfeller. Imidlertid er rekkevidden av forskjellige mutasjoner I aloxe3-genet begrenset, på grunn av overvekt av 2 mutasjoner, s.Arg234X og p.Pro630Leu, som synes å svare til hotspots.37,74,75
pasientene med mutasjoner I aloxe3-og ALOX12B-genene viser vanligvis EN CIE-fenotype.74,75,77 alvorlighetsgraden av skalering er mild eller moderat, og skalaene har en hvitaktig eller lysebrun farge. Erytem kan også være tilstede. Så mange som 76% av pasientene er født som collodion babyer og 88% har svetteforstyrrelser.37 Pasienter med mutasjoner I alox12b-genet viser mer begrenset, hvitaktig avskalling sammenlignet med bærere av mutasjoner I aloxe3-genet. I disse tilfellene er skalaene brune og vedhengende. Tilstedeværelsen av erytem, palmoplantar hyperkeratose og accentuering av palmoplantar-foldene er også forbundet MED alox12b mutasjoner.37
Ichthyin / NIPAL4
nipal4-genet, også kjent som ichthyin-genet, ligger på kromosom 5q33. Den har 6 eksoner som koder for et protein med flere transmembrane domener av ukjent funksjon.84 det er antatt at proteinproduktet deltar i samme metabolismevei som LOX og kan virke som en reseptor for trioksiliner A3 Og B3 eller for andre metabolitter av hepoksilinmetabolismevei.84 Det ville således være involvert i dannelsen av lamellære legemer eller i deres transport mot det ekstracellulære rommet.32 til støtte for dette er 2 observasjoner. For det første, i 93% av tilfellene, mutasjoner i dette genet er forbundet med ultra mønster medfødt iktyose type 3, karakterisert ved abnormiteter i lamellære legemer og tilstedeværelsen av langstrakte perinukleære membraner i stratum granulosum.32 Sekund, nipal4 uttrykkes hovedsakelig i stratum granulosum av epidermis, hvor lamellarlegemene er tilstede.85
Siden oppdagelsen AV nipal4-genet i 2004,84 er det kun rapportert 9 mutasjoner hos pasienter Fra Middelhavslandene (Algerie, Tyrkia Og Syria), 84 Skandinaviske land,32 Pakistan,85 Færøyene, 32 Og Sør-Amerika.84
det kliniske spekteret av pasienter med mutasjoner i dette genet er bredt, selv blant medlemmer av samme familie. Mellom 3,7% 32 og 60% 84 er født som collodion babyer. Når kollodionmembranen forsvinner, utvikler de fleste pasienter manifestasjoner AV CIE, med fine hvite vekter på en erytematøs base på ansikt og trunk og større, brune vekter på nakke, rumpe og ben.84 Markert xerose, generaliserte brune retikulære hyperkeratotiske plakk som vises fremhevet i hudfoldene, og ansiktsdyschromi kan være tilstede.32,85 i tillegg er palmoplantar keratoderma et hyppig funn sammen med sporadiske fingerkontrakter og buede fingernegler. Noen studier har rapportert funn som er mer typiske FOR LI.32,85 tilstedeværelse av tegn og symptomer på atopisk dermatitt er rapportert hos noen pasienter, selv om mutasjoner I flg-genet ikke ble påvist i noen av disse tilfellene.85
CYP4F22
flj39501-eller CYP4F22-genet ligger på kromosom 19p13.12.86 det har 12 eksons87 og koder for en p450 cytokrom, familie 4, underfamilie F, polypeptid 2, homolog for leukotrien B4-ω-hydroksylase (CYP4F2). Reaksjonen som katalyseres AV PRODUKTET AV FLJ39501 i huden og substratene til denne reaksjonen kan utledes ved analogi med de kjente HOMOLOGENE CYP4F2 OG CYP4F3.88 det er antatt AT CYP4F2 og CYP4F3 deltar i hepoksilinveien ved å katalysere omdannelsen av trioksilin A3 til 20-hydroksy-(r)trioksilin A387, og at sluttproduktet av DENNE veien, 20-karboksy-trioksilin A3, kan ha en viktig biologisk regulerende effekt på huden.89
hittil har bare 8 mutasjoner av dette genet blitt rapportert i 12 consanguineous familier Fra Middelhavslandene 87 og i 1 familie Av Israelsk opprinnelse.62
i familiene rapportert Av Lef@vre et al., 87 de fleste pasientene hadde EN CIE-fenotype ved fødselen, og Dette utviklet seg senere TIL LI. Pasienter ble vanligvis født med merket erytroderma, men uten kollodionmembran. Som de ble eldre, de utviklet generalisert hvitaktig-grå skalering, som var mer markert i periumbilical regionen, på baken, og på den nedre delen av kroppen. Hyperlinearitet av palmer og såler og desquamation i hodebunnen, til tider av pityriasiform type, var hyppig.87 i en annen familie ble de 3 berørte medlemmene født som collidion babyer og utviklet intens erythroderma, generalisert desquamation og palmoplantar keratoderma.62
ABCA12
I 2003 BLE abca12-genet rapportert å være ansvarlig for noen TILFELLER AV LI og ble kartlagt til kromosom 2q34.4.det ble senere bekreftet at mutasjoner i dette genet også var ansvarlige for HI.2, 3abca12 koder 53 eksoner, og tilhører en familie AV ABC-transportører, som binder adenosintrifosfat samtidig som det letter transporten av flere molekyler over cellemembranen.90 medlemmene AV ABCA-underfamilien er alle involvert i lipidtransport.91 Mangelfull ABCA12-funksjon forårsaker lipidtransportforstyrrelser i lamellare legemer og fører dermed til en reduksjon i intercellulære lipidnivåer i stratum corneum.3ultrastrukturelle studier har vist AT ABCA12 er lokalisert i lamellare legemer assosiert med glykosylceramider.91abca12 mutasjoner har vært assosiert med forstyrrelser i distribusjon og transport av glykosylceramider og med reduserte nivåer av hydroksykeramider, en av hovedkomponentene i lipidbarrieren i de intercellulære rom.3,6,92,93 den massive hyperkeratosen som oppstår hos disse pasientene, kan være en kompenserende respons på en mangelfull lipidbarriere.94 Det kan også skyldes mangel på desquamation av corneocytes, 93 som kan være forårsaket av defekter i transport av visse proteaser, som callicrein 5 og cathepsin D, som følge av forstyrrelser i lamellar legemer.95 Murine modeller og in vitro studier tyder PÅ AT ABCA12 mutasjoner også har en effekt på epidermal differensiering.95-97
til dags dato har mer enn 50 mutasjoner blitt rapportert I abca12-genet hos pasienter MED ARCI Fra Afrika, Europa, Pakistan og Japan. De hyppigste mutasjonene er p.Val244SerfsTer28,2,98,99 identifisert I Pakistanske og Indiske populasjoner, og P.Asn1380ser, 4 identifisert I Afrikanske familier. I begge tilfeller kan disse være grunnleggende mutasjoner.
omfanget AV ABCA12-mutasjonene er relatert til fenotype, med mutasjoner assosiert med fullstendig tap av funksjon som fører TIL HI-fenotypen.2,3,98 – 102 Derimot, I LI og CIE, er de fleste mutasjoner missense, og har en mindre alvorlig effekt på proteinfunksjonen.4-6, 103 mutasjonene som ligger til GRUNN FOR li-fenotypen synes å være konsentrert i det første adenosintrifosfatbindende kassettområdet.4 Klinisk har pasienter Med CIE og mutasjoner I abca12-genet mellomstore skalaer som er noe større enn de som vanligvis observeres hos pasienter med denne fenotypen.
HARLEQUIN Iktyose
HI eller HARLEQUIN fetus er en alvorlig og vanligvis dødelig form for iktyose. Barnene er vanligvis for tidlige med omfattende skinnende hyperkeratotiske plakk, skilt av dype sprekker, som dekker hele integumentet og danner geometriske mønstre som minner om klær som bæres av harlequins, og gir dermed tilstanden sitt navn. Tetthet i huden fører til markert vridning av øyelokk og lepper, rudimentær utvikling av ledd og nesebrusk og noen ganger mikrocefali. Barnene har sjelden øyenvipper eller øyenbryn, selv om håret i hodebunnen kan bli bevart. Hendene og føttene er hovne og edematøse, og ofte dekket av et hanskelignende lag. De kan ha fingerkontrakter.
for slike pasienter er risikoen for å dø i nyfødtperioden svært høy.104 Lungeventilasjon er kompromittert; transepidermalt vanntap fører til dehydrering, vannkraftubalanse og termisk ustabilitet; og risikoen for infeksjoner øker. Tetthet i ansiktet og eclabium hindrer suging og dermed mating, med tilsvarende forverring av dehydrering. Nyfødte med denne tilstanden levde sjelden lenger noen uker. I de senere år har imidlertid sjansene for langsiktig overlevelse økt betydelig, hovedsakelig på grunn av administrasjon av systemiske retinoider og fremgang i intensiv neonatal omsorg.105 i en nylig studie overlevde 83% av pasientene behandlet med orale retinoider sammenlignet med 24% av ubehandlede pasienter. De fleste dødsfallene skjedde i de første 3 dagene av livet, men behandlingen ble ikke startet før etter dette hos mange av de overlevende.104 Dette ville tyde på at mange av disse tidlige dødsfallene ville ha skjedd uavhengig av retinoidbehandling.
barna som overlever nyfødtperioden utvikler generelt alvorlig CIE.106 arten og plasseringen av mutasjoner I abca12-genet og omfanget av tap av transportørfunksjon kan bestemme prognosen.3,92,107 Pasienter som sparer en viss grad av proteinaktivitet, om enn minimal, kan ha en bedre sjanse til å overleve. Bærere av homozygote mutasjoner har høyere dødelighet.104
DEN viktigste histologiske egenskapen TIL HI er tilstedeværelsen av et ekstremt tykt og kompakt orthokeratotisk stratum corneum. Hårsekkene og svettekanalene har fremtredende hyperkeratotiske plugger 107, 108 og har unormale eller fraværende lamellære legemer, lipidinneslutninger eller rester av organeller eller kjerner i corneocytene, og fravær av intercellulære lipider i den ultrastrukturelle studien.108,109 hårsekkene viser en markert konsentrasjon av keratotisk materiale, som er et diagnostisk trekk VED HI som brukes til prenatal diagnose.
hittil er deteksjonshastigheten for mutasjoner I abca12-genet hos PASIENTER med HI nær 100%, og dette synes derfor å være en genetisk homogen tilstand.
Collodion Baby Og Self-healing Collodion Baby
Collodion babyer er vanligvis født for tidlig og perinatal sykelighet og dødelighet er økt. Ved fødselen er den nyfødte dekket av en skinnende undervist gjennomsiktig membran som minner om cellofaninnpakning (Fig. 5). Babyene har ectropion, eclabium og hypoplasi i nese-og leddbrusk. Suging og lungeventilasjon kan hindres110 og transepidermalt tap av vann og risikoen for infeksjoner øker.110,111

Collodion baby som senere utviklet seg til en lamellær iktyose fenotype.
Collodion baby er den vanlige presentasjonen FOR HI og CIE. Autosomal dominant LI,112,113 Sjö-Larssons syndrom, 110 trichothyodystrofi, 114 Juvenil Gauchers sykdom, 110 nøytral lipidlagringssykdom, Conradi-Hü-Happle syndrom, Hays-Wells syndrom og ektodermal dysplasi115 kan også av og til forekomme som collodion baby. Membranen forsvinner spontant i 10% til 24% av nyfødte, for å gi vei til helt normal hud.110.116 tidligere ble disse tilfellene beskrevet SOM li av nyfødte, 117, men DE er ikke referert TIL SOM SHCB.118 noen forfattere har foreslått begrepet selvforbedrende kollodion iktyose fordi mange av disse pasientene, når de ble undersøkt senere i barndommen eller som voksne, har en variabel grad av anhidrose og varmeintoleranse og milde tegn på iktyose, som xerose og fin desquamation, spesielt i axillae og nakke.78
verken optisk mikroskopi eller ultrastrukturelle undersøkelser av collodion baby er spesifikke. Det er derfor å foretrekke å utsette hudbiopsien til den endelige fenotypen har utviklet seg.
Mutasjoner I tgm1,7,119alokse3,78 og ALOKS12B23,78,79 gener har blitt identifisert hos pasienter med SHCB. ALOX12B mutasjoner er de vanligste. I en serie På 15 Skandinaviske pasienter med SHCB hadde 67% mutasjoner I alox12b-genet, 25% I ALOXE3-genet og 8,3% i TGM1-genet.78 Mutasjoner ble ikke funnet hos noen pasienter, og så andre gener er også sannsynlig å bli involvert. Det har vært spekulasjoner om at disse mutasjonene reduserer enzymatisk aktivitet i livmoren, men ikke etter fødselen.7 i livmoren, hvor det hydrostatiske trykket er høyt, omdanner chelation med vann det muterte enzymet til en inaktiv konformasjon. Etter fødselen, når trykket avtar, går enzymet tilbake til sin aktive form og aktiviteten øker tilstrekkelig til å opprettholde en normal eller minimal påvirket fenotype.7
Acral Selvhelbredende Collodion Baby
selv om collodion baby påvirker hele kroppen, har tilfeller begrenset til acral regioner blitt rapportert. I 1952, Finlay et al.120 rapporterte et tilfelle av kollodionmembran som bare påvirket hender og føtter, og som fulgte et selvhelbredende kurs. Nylig har et nytt tilfelle av acral SHCB blitt rapportert i forbindelse med mutasjoner AV tgm1-genet.8 det er ikke kjent hvorfor disse lesjonene er begrenset til akralregioner, selv om faktorer forbundet med stedsavhengig regulering av enzymaktivitet kan være i drift.8
Badedrakt Iktyose
badedrakt iktyose ble først rapportert som en uavhengig ARCI-variant i 2005, selv om tilfeller av iktyose med en spesiell fordeling hadde blitt rapportert tidligere.121-123 Det har blitt påvist hovedsakelig hos pasienter Av Sørafrikansk opprinnelse, 9 selv om det også er rapportert hos personer Fra Europa og Middelhavslandene.124 ved fødselen har pasienter en generalisert kollodionmembran som deretter kaster for å forlate den karakteristiske fordeling av skalering. Stammen, den proksimale delen av armene, inkludert axillene, nakken og hodebunnen, påvirkes vanligvis, mens den sentrale delen av ansiktet, lemmer og binyrene er vanligvis spart.9 vekten er stor, lamellær og mørk i fargen. Finere desquamation kan forekomme i popliteal og antecubital fossae.124,125 håndflatene og fotsålene har mild diffus hyperkeratose mens ryggen på hender og føtter viser ingen involvering.
Histopatologisk studie av berørt hud viser markert hyperkeratose uten parakeratose, normale granulære lag, mild eller moderat akanthose og et mildt lymfocytisk infiltrat i øvre dermis.9 Elektronmikroskopiobservasjoner er i samsvar med medfødt iktyose type 2 i de fleste tilfeller. Uinvolvert hud viser ingen unormale funn.124,125 i sunn hud Er TGase 1-aktivitet litt redusert og vanligvis lokalisert i perikellulære områder. I involvert hud er enzymatisk aktivitet gjenværende og unormalt lokalisert i cytoplasma.124
Mutasjoner har blitt påvist I tgm1-genet hos alle pasienter med badedrakt iktyose studert til dags dato.119,124 – 126 den vanligste mutasjonen er P. Arg315Leu, som har blitt identifisert hos De Fleste Sørafrikanske pasienter og kan være en grunnleggende mutasjon. Oji et al.124 foreslo at hudtemperatur kan spille en rolle i utviklingen av disse manifestasjonene. Ved hjelp av digital termografi viste forfatterne en sterk sammenheng mellom kroppstemperatur og desquamation, med de varmeste områdene i kroppen som de mest berørte. Aufenvenne et al.127 viste en reduksjon i optimal temperatur For TGase 1-aktivitet hos pasienter med badedrakt iktyose. Denne reduksjonen ble ikke observert hos friske kontroller eller hos pasienter med generalisert LI. denne reduksjonen i temperatur ville forklare fenotypen til disse pasientene. Den optimale temperaturen er 37 hryvnias C For det normale enzymet, men 31 hryvnias C for det muterte enzymet.
Behandling
hovedformålet med behandling i iktyose er å eliminere skalering og redusere xerose uten å forårsake overdreven irritasjon (Tabell 3). Før man bestemmer seg for behandling, bør aspekter som pasientens alder og kjønn, type og alvorlighetsgrad av sykdommen, og omfang og sted for lesjonene tas i betraktning.128
Terapeutisk Strategi I Autosomal Recessiv Medfødt Ichthyoses.
| Terapeutisk strategi for autosomal recessiv medfødte ichthyoses | |
| Bading og mekanisk eliminering av skalaer | Bading med natriumbikarbonat eller hvetestivelse, maisstivelse eller risstivelse; mekanisk fjerning av skalaene (1 eller 2 ganger om dagen) |
| Topisk behandling (sekvensiell) | ureaholdige fuktighetskremerkeratinolytika med propylenglykolkombinert keratinolytika (propylenglykol, α-hydroksysyrer eller urea)Keratinolytika kombinert med salisylsyreaktuelt retinoidsIn nyfødte og små barn, påfør et kjøretøy uten aktive ingredienser. Unngå urea, salisylsyre og melkesyre på grunn av risiko for systemisk absorpsjon |
| Oral behandling | Orale retinoider (acitretin eller isotretinoin) |
| Andre tiltak | oppfølging av ektropion ved oftalmologenvanlig rensing av ytre øret ved øre-hals-nese spesialistfysioterapi for å forhindre kontrakturer.Unngåelse av anstrengende aktiviteter i en høy omgivelsestemperaturhydroterapi |
Bading Og Mekanisk Eliminering Av Skalaer
Daglig bading anbefales for pasienter MED ARCI å mekanisk eliminere skalaer og spor av fuktighetskrem. Dette er lettere hvis pasienten er nedsenket i vann i 15 til 30minutter. Noen forfattere anbefaler å legge natriumbikarbonat til badet for å denaturalisere keratinene og gjøre vannet alkalisk, og dermed lette eliminering av skalaene.129 andre produkter som kan tilsettes inkluderer hvetestivelse, maisstivelse eller risstivelse. Badeoljer er ikke egnet da de kan føre til okklusjon med påfølgende risiko for bakteriell proliferasjon og forverring av termoregulering.
Aktuell Behandling
Fuktighetskremer og aktuelle keratolytiske midler er vanligvis det første terapeutiske alternativet. De forbedrer hudens barrierefunksjon og letter avskalling. Milde lokale bivirkninger, som forbigående kløe, irritasjon eller stikkende følelse kan forekomme.
Natriumklorid, urea, vitamin e acetat, glyserol og vaselin kan brukes som fuktighetskremer og smøremidler. Hos pasienter med tykk skalering og markert hyperkeratose kan 1 eller flere keratolytiske midler, som α-hydroksysyrer (melkesyre og glykolsyre), 130 salisylsyre,n-acetylcystein, 131-133 urea (>5%), 134 og propylenglykol,tilsettes. Modulatorer av keratinocytdifferensiering brukes også. Disse inkluderer aktuelle retinoider (tretinoin, adapalen, tazaroten),135,136 kalsipotriol,137 og dexpanthenol.Aktuelle retinoider forårsaker ofte irritasjon og små, svært smertefulle sprekker.137 Videre er det risiko for absorpsjon og teratogenitet hos fertile kvinner hvis de brukes for mye.138 for å øke effektiviteten av keratolytika og fuktighetskrem, kan okklusiv dressing påføres i bestemte områder ildfast til behandling.139 en additiv eller synergistisk effekt kan også oppnås ved å kombinere 2 eller flere keratolytiske midler eller fuktighetskrem.140-142 Behandling bør optimaliseres for hver enkelt, gitt den svært variable karakteren av tilstanden og hudfølsomhet og forskjeller i respons på hver behandling. Optimaliseringsprosessen kan bli hjulpet ved å behandle den ene siden av kroppen annerledes enn den andre for å muliggjøre sammenligninger. Nyfødte og små barn bør behandles med et kjøretøy uten noen aktive stoffer, da huden er veldig fin og følsom, og de fleste keratolytika tolereres ikke. I tillegg er risikoen for perkutan absorpsjon av aktuelle produkter som urea, salisylsyre og melkesyre større.143-145
Systemisk Behandling
Orale retinoider har keratolytiske effekter som bidrar til å eliminere skalaer og forhindre overdreven hyperkeratose. Både isotretinoin og aromatiske retinoider (acitretin og etretinat) har vist seg å være effektive i behandlingen Av ARCIs.128.146.147 Acitretin i en dose på 0,5 til 1 mg / kg / d er det mest brukte stoffet, spesielt hos PASIENTER MED LI. 148 Pasienter MED CIE kan ha en mer fullstendig respons og ved lavere doser.
de viktigste bivirkningene er mukokutane lidelser, teratogenisitet, muskel-og skjelettlidelser og unormal lipidprofil og transaminaseøkning.149-152 med hensyn til teratogenisitet, i tilfelle av etretinat og acitretin, bør legemidlene unngås under graviditet, og pasienter bør unngå å bli gravid i 3 år etter seponering av behandlingen.151 Isotretinoin har kortere halveringstid og elimineres helt fra organismen etter 1 måned, og kan derfor være det foretrukne alternativet hos kvinner som ønsker å bli gravide.128
behandlingsovervåking bør omfatte laboratoriearbeid med leverfunksjonstest og lipidprofil før behandlingsstart, deretter 1 måned og hver 3. måned etter behandlingsstart. Hos fertile kvinner bør en graviditetstest utføres i 2 uker før behandlingsstart, og et effektivt prevensjonsmiddel bør brukes fra 4 uker før behandling til 3 år etterpå (i tilfelle av acitretin). Når langvarig behandling er nødvendig med retinoider, bør vekst og beinutvikling overvåkes. Noen forfattere foreslår å utføre en beinstudie før behandling etterfulgt av en årlig undersøkelse.151 Nyere retningslinjer anbefaler ikke å utføre rutinemessig radiografi på grunn av mulige skadelige effekter.152 i Stedet anbefales selektive radiografiske studier hos pasienter som har atypisk bein smerte.152
et alternativ til systemisk retinoidbehandling er bruk av legemidler kjent som retinsyre metabolisme blokkerende midler, som øker de endogene nivåene av retinsyre. Et slikt legemiddel er liarozol, som har fått foreldreløs status for behandling AV LI, CIE og HI av European Medicines Agency og US Food And Drug Administration.153-155 dette stoffet har vist seg å være mer effektivt enn acitretin i kliniske studier, og det tolereres også bedre og har en bedre farmakokinetisk profil.154
Annen Medisinsk Behandling
hos pasienter Med ektropion kan påføring av kunstige tårer og øyesmøremidler og fuktighetsgivende huden i ansiktet og kinnene spesielt redusere palpebral tilbaketrekning. Kirurgisk korreksjon er et gyldig alternativ i alvorlige tilfeller, men dette må vanligvis gjentas noen år senere. Hydroterapi kan være gunstig.156 Pasienter bør rådes til å unngå anstrengende fysisk aktivitet når omgivelsestemperaturen er høy, da hypohidrose medfører risiko for varmeslag og kramper. Orale retinoider kan forbedre termoreguleringen.157 Fysioterapi er viktig for å forhindre fleksjonskontraktur, spesielt NÅR DET GJELDER HI. Regelmessig rensing av den eksterne hørskanalen av en øre-hals-nese-spesialist kan forhindre skalaer i å samle seg og forhindre hørselstap.
Genetisk Rådgivning Og Fosterdiagnostikk
når en pasient er diagnostisert Med iktyose, bør han eller hun bli tilbudt passende genetisk rådgivning der arten av uorden, overføringsmodus, og risikoen for fremtidige manifestasjoner i familien er forklart. Prenatal diagnose kan indikere om fosteret er berørt, og hvis dette er tilfelle, kan psykologisk forberedelse av familien tilbys og problemer som forventes under graviditet og fødsel. Foreldrene kan få mulighet til abort hvis ingen behandling er tilgjengelig. I tillegg, hvis genterapi for disse forholdene blir tilgjengelig i fremtiden, vil prenatal diagnose muliggjøre anvendelse av denne terapien så tidlig som mulig.
i mer enn 20 år ble prenatal diagnose utført ved å ta en biopsiprøve av føtal hud og studere den ved optisk mikroskopi, elektronmikroskopi eller immunhistokjemi.158,159 denne invasive prosedyren kunne bare utføres i de sene faser av svangerskapet, mellom uke 15 og 23 av svangerskapet, og var forbundet med en 1% til 3% risiko for å miste fosteret.160,161 identifisering av molekylære mekanismer av arvelige hudsykdommer har muliggjort en mye tidligere diagnose basert på genetiske teknikker.102,162-164 Foster DNA er oppnådd ved amniocentese utført mellom uke 15 og 20 eller ved chorionisk villusprøvetaking mellom uke 10 og 12. Risikoen for fostertap med disse teknikkene er mindre enn mellom 0,5% og 1%.165 andre ikke-invasive metoder i utvikling er analyse av føtal celle DNA og fritt føtal DNA i mors sirkulasjon166 samt bruk av 3-dimensjonal ultralyd.167,168
Preimplantasjonsgenetisk diagnose kan også være mulig i in vitro befruktningsteknikker, slik at bare befruktede egg uten mutasjon blir implantert i livmoren, og dermed unngår behovet for abort i de fleste tilfeller.169
Fremtidige Strategier For Genetisk Behandling Av Iktyose
selv om det er gjort viktige fremskritt i den genetiske diagnosen iktyose, blir også nye strategier forfulgt for disse sykdommene.170 huden er det mest tilgjengelige organet for genoverføringsterapi, og slike teknikker er derfor minimalt invasive.171 men huden har også unike immunologiske egenskaper som ikke favoriserer langsiktig uttrykk for et transgen produkt.172 I LI klarte en prosess med ex vivo genoverføring å gjenopprette normalt tgm1-uttrykk og korrigere fenotypen av hudtransplantert på baksiden av immunsupprimerte mus.173,174 nylig har fenotypen av dyrkede keratinocytter fra PASIENTER MED HI på grunn AV mutasjoner I abca12-genet også blitt gjenfunnet.3
Interessekonflikter
forfatterne erklærer at de ikke har noen interessekonflikter.