Frontiers in Neurology
Innledning
Neuroacanthocytosis Er en overordnet betegnelse for en gruppe sjeldne syndromer som er preget av nevrologiske symptomer i kombinasjon med spiky deformerte røde blodlegemer (acanthocytes). Denne rapporten fokuserer på Chorea-acanthocytosis (ChAc), som er en foreldreløs sykdom med anslagsvis 1000 berørte individer over hele verden forårsaket av autosomal-recessive mutasjoner I vps13a (vacuolar protein sortering 13 homolog a) genet på kromosom 9q (1). Sykdommen går en progressiv kurs, årsaker til økt dødelighet kan bli avvist motorfunksjoner korrelert med risikotilstander som dysfagi, men også plutselige uventede dødsfall er beskrevet (2, 3). Så langt er det ingen årsaksbehandling.
den mistenkelige sameksistensen av akanthocytose og bevegelsesforstyrrelser ble først rapportert På 1970-tallet Av Levine og Critchley (4, 5) og har siden blitt akseptert som det fremtredende trekk ved sykdommen. Men med vår saksrapport beskriver vi en sjelden klinisk fenotype Av ChAc som mangler åpenbare bevegelsesforstyrrelser, noe som tyder på et bredere utvalg av kliniske presentasjoner. Diagnostiske verktøy må møte disse utfordringene for å etablere riktig diagnose, vi foreslår her molekylær neuroimaging som en nøkkelmetode.
Saksrapport
den mannlige indekspasienten ble først presentert ved 25 års alder med to uprovoserte bilaterale tonisk kloniske anfall. Det var ingen medisinsk historie. Hjernen MR-skanning og EEG i den alderen var normale og ingen behandling ble startet på grunn av pasientens motvilje og sjeldne hendelser. Imidlertid hadde pasienten pågående anfall som nå presenterer som delvis epilepsi. Han beskrev reoccurring følelser av plutselig svimmelhet som ble diagnostisert som en vertiginous aura. Videre hadde han dyskognitive anfall der han viste enten (a) manglende respons og resitering av tyrkiske bønner, (b) oral automatisme, eller (c) fiddling med hendene og uttering av lyder—alt utviklet seg delvis til tonisk klonisk anfall. Video-EEG viste nå lokale spike-bølge komplekser både i høyre og venstre tinninglappen. I samsvar med semiologien av anfall ble diagnosen bilateral temporal lobe epilepsi gjort og medisinering med levetiracetam ble startet.
i en alder av 31 anfall ble ikke fullstendig undertrykt, som en del av videre diagnostisering av temporal lobe epilepsi, gjennomgikk pasienten EN FDG-PET (Figur 1) som viste en bilateral mesiotemporal hypometabolisme, i tråd med mesiotemporal anfallsopprinnelse. Det var imidlertid også en markert, svært uvanlig striatal hypometabolisme som hevet mistanke om en nevrodegenerativ bevegelsesforstyrrelse. Det ble derfor igangsatt en omfattende oppfølgingsevaluering (Se Figur 2). EN FP-CIT-SPECT ble utført som ga et moderat bilateralt tap av striatal dopamintransportørtilgjengelighet, i tråd med en nedgang i nigrostriatal integritet (Figur 1). En høyoppløselig MR presenterte nå bilateral atrofi av kjernen caudatus (Figur 3). Nevrologisk undersøkelse viste et diskret bundet gangmønster og hypomimi, men ingen annen affeksjon av det ekstrapyramidale motorsystemet og ingen bulbar symptomer som å mate dystoni. Bortsett fra lav refleksstatus på nedre lemmer, hvor nerveledningsstudier bekreftet en sensorimotorisk aksonal polyneuropati, var nevrologisk undersøkelse normal. Nevropsykologiske symptomer inkluderte aspekter av en angstlidelse, og pasienten rapporterte vanskeligheter med korttidshukommelsestap. Neuropsykologisk testing bekreftet imidlertid ikke manifest mnestic funksjonsfeil, men heller en uspesifikk distraksjon, som i tillegg kunne ha blitt forsterket av utilstrekkelig anfallssuppresjon på den tiden. Laboratorietesting var negativ for symptomatisk epilepsi (dvs. antistoffer, antineuronale antistoffer). Cerebrospinalvæske viste ingen tegn på betennelse, men en moderat proteinøkning (765 mg / l). Analyse av biogene aminer i cerebrospinalvæsken viste en forhøyelse av glutamin som vi assosierte med nylige anfall. I perifert blod ble 0,4% av akanthocytter funnet. Serologisk testing for Wilsons sykdom var negativ. Merkbar var en vedvarende økning av kreatinkinase (område: 1000-3000 U / l) som førte til den mistenkte diagnosen av en mitokondriell lidelse. For videre undersøkelser ble det utført en iskemisk laktatprøve som viste normale resultater. En muskelbiopsi Av m. gastrocnemicus ble utført, men analyse av mitokondriell funksjon og respiratorisk kjede var normal.
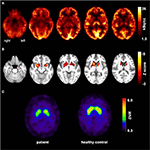
Figur 1. Resultater av molekylær imaging studier. Transaksiale fdg-PET-bilder (A) viste ikke bare en mild bilateral mesiotemporal hypometabolisme (i tråd med mesiotemporal anfallsopprinnelse), men også en markert striatal hypometabolisme som ble funnet å være svært signifikant sammenlignet med friske kontroller . En ytterligere fp-CIT-SPECT-undersøkelse (C) viste også et moderat tap av striatale dopamintransportører, og opplevde en nedgang i nigrostriatal integritet(en aldersmatchet sunn kontroll er vist for sammenligning; DVR, distribusjonsvolumforhold).

Figur 2. Klinisk og diagnostisk kurs på indeksen pasienten.
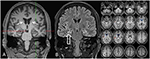
Figur 3. MR viser diskret bilateral kaudat hodeatrofi og en mindre og hyperintense høyresidig hippocampus som indikerer høyresidig hippocampal sklerose . Den venstre hippocampus er noe hyperintense, men ikke atrofisk. Bilateral kaudat hodet atrofi er bekreftet MED cvr analyse, der blå farger viser redusert grå og røde farger økt cerebrospinalvæske volum, henholdsvis (C).
Familiehistorie avslørte en slektskap med sine foreldre (søskenbarn), som selv ikke hadde noen signifikant medisinsk historie. Av hans seks søsken hadde to (mellom 20-30 år) nå også lidd av generelle anfall (Se Figur 4). Medisinske poster av hans søsken var ikke tilgjengelig for oss.
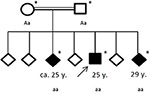
Figur 4. Stamtavle av den berørte familien, med alder av første epileptisk anfall i år (y). Arrow: indeks patent. Asterisk: genetisk testing utført. Aa, heterozygot; aa, homozygot for c.4326 T> A (S. Tyr1442*).
under hypotesen om en arvelig nevrodegenerativ bevegelsesforstyrrelse ble pasienten og hans familie henvist til genetisk testing. Exome sekvensering avslørte en homozygot roman avkortende mutasjon c.4326 T> A (P. Tyr1442*) I chac genet VPS13A i de tre berørte søsken. De consanguine foreldrene ble begge oppdaget heterozygote. Ingen andre varianter eller mutasjoner ble identifisert i exome-sekvenseringen.
faktisk, i en oppfølging i en alder av 33 år, hadde indekspasienten fortsatt bare marginale ekstrapyramidale symptomer, og ingen var tilstede hos noen av hans søstre. Han ga nå inntrykk av liten stivhet bare under priming på høyre øvre del, og en mild bradykinesi i øvre lemmer. Under behandling med levetiracetam og lakosamid har pasienten vært anfallsfri.
Diskusjon
Med den foreliggende sak, viser Vi at ChAc kan klinisk manifestere med temporal lobe epilepsi mangler noen signifikante motoriske symptomer på grunn av en ny tilsvarende mutasjon I vps13a genet.
sykdomsforløpet er typisk preget av en progressiv bevegelsesforstyrrelse (inkludert chorea, dystoni, parkinsonisme), kognitive og psykiatriske endringer og myopatiske symptomer med serologiske markører for akanthocytose og hyperkemi. Beslag har tidligere blitt notert som et symptom På ChAc (6), men bevegelsesforstyrrelser anses fortsatt som nøkkelsymptomet. Bevis oppstår at epilepsi, og mer spesifikt epilepsi med opprinnelse i tinninglappen, kan være en undervurdert fenotype Av ChAc. Peluso et al. rapportert en pasient som ligner på vår, som, selv i en alder av 46 år, ikke viste bevegelsesforstyrrelser mens de presenterte neuroimaging funn som tyder på basal ganglia involvering (7). I samsvar Med Det, Scheid et al. beskrevet tre pasienter som led av mesial temporal lobe epilepsi og sklerose (basert PÅ MR-studier) som det dominerende symptomet og ble diagnostisert Med ChAc (8). Videre Mente et al. nylig gitt obduksjonsresultater Av En ChAc pasient som viste ikke bare basalganglia atrofi, men også hippocampal sklerose (9). I tråd med vårt tilfelle synes bilateral involvering av hippocampus å være et vanlig funn (10, 11). Det nøyaktige forholdet mellom anfall og sklerose er imidlertid fortsatt et spørsmål om høne eller egg. Choreins korrelerende rolle i den strukturelle utviklingen av hippocampus er ennå ikke fullt ut forstått. Interessant, i en musemodell Av chac strukturelle endringer ble sett, som hippocampal protein ekspresjon AV GABA (A) reseptor gamma 2 og dens forankring protein chorein ble økt (12).
i klinisk rutine kan denne fenotypen fortsatt være utfordrende for å finne riktig diagnose. Det første avgjørende hintet mot en nevrodegenerativ sykdom hos pasienten var en fremtredende nedgang i striatal glukosemetabolisme og en moderat nedgang i nigrostriatal integritet på henholdsvis fdg-PET og FP-CIT-SPECT. Den lille samlingen av funksjonelle avbildningsresultater, basert på case seriebeskrivelse, viser at endringer i metabolisme og dopaminerg dysfunksjon hovedsakelig forekommer i kaudatkjernen og putamen (13). HJERNEMRI avslørte atrofi av kjernen caudatus i løpet av sykdommen, som har blitt beskrevet som et typisk funn i ChAc (14). Følgelig inkluderer distinkte nevrologiske egenskaper vanligvis chorea, mating dystoni, orofaciolingual dyskinesier og tics, sammen med andre symptomer på basal ganglia kjærlighet som parkinsonisme og dystoni (3). Det er fristende å spekulere på at de observerte striatale endringene hos pasienten fortsatt var under terskelen for å forårsake symptomer (dvs.prodromale bildefunn), som til slutt kan oppstå etter hvert som sykdommen utvikler seg. Vi oppfordrer derfor strukturell og molekylær avbildning som et sensitivt diagnostisk verktøy i denne foreldreløse sykdommen.
i ChAc er det beskrivelser av acanthocyttall mellom 5 og 50% (3), men det er noen kasusrapporter som viser at acanthocytes bare kan vises sent i sykdomsforløpet (15), kan være svært lave, eller til og med være helt fraværende (16). Derfor bør acanthocytter ikke anses som obligatoriske for å gjøre diagnosen. I tillegg epilepsi kan forsinke diagnose som det skjuler andre typiske symptomer På ChAc. Spesielt kan psykiatriske symptomer som personlighetsendringer og kognitiv svekkelse, som er svært vanlige, feiltolkes og tilskrives legemiddelrelaterte bivirkninger (dvs.av levetiracetam) eller anfallsrelaterte. Sekundær hyperkemi er sett etter generelle tonisk kloniske anfall og en vedvarende høyde kan overses.
vårt tilfelle fremhever også den avgjørende fordelen med exome-sekvensering for å etablere en korrekt diagnose, spesielt i sjeldne sykdommer eller når symptomene er vage. Familien av pattedyrs vps13 (A-D) – gener er av økende interesse og mutasjoner er identifisert i andre nevrologiske sykdommer som autosomal recessiv Parkinsons sykdom eller autosomal recessiv spinocerebellar ataksi (17). Recessive mutasjoner i VPS13D forårsaker bevegelsesforstyrrelser i barndommen (18). Den underliggende patofysiologien Til ChAc er ennå ikke fullstendig dechiffrert, men DET ble vist AT VPS13A koder proteinkoreinet som uttrykkes allestedsnærværende i hjernen og i et stort antall andre vev (19). Studier viser at det deltar i signalveier som regulerer cytoskeletal arkitektur, eksocytose og celleoverlevelse (20). En annen mutasjon som er beskrevet å presentere med en anfallsdominert fenotype hos 9 pasienter Med ChAc er c.2343del-mutasjonen I vps13a-genet (21). Det er rimelig å anta at spesifikke mutasjoner i samme gen resulterer i en viss aberrasjon av korein som forårsaker en unik fenotype, for eksempel ved å påvirke forskjellige områder av hjernen. Imidlertid er den underliggende patofysiologien fortsatt bare spekulativ og ytterligere dokumentasjon av de forårsakende mutasjonene vil være av interesse. Vi viser her at den avkortende mutasjonen c. 4326 T> A (P. Tyr1442*), som ikke er beskrevet tidligere, resulterer i en lignende fenotype med dominerende epilepsi hos alle berørte familiemedlemmer.
Avsluttende Bemerkninger
Epilepsi (spesielt bilateral temporal lobe epilepsi) er sannsynligvis en dominerende fenotype Av ChAc. Derfor bør et søk etter flere røde flagg (som hyperkemi, familiehistorie, polyneuropati) utføres nøye. I mistenkelige tilfeller søk etter acanthocytes i blod OG hjerne MR bør legges som disse verktøyene er allment tilgjengelig. Hvis du er i tvil diagnostikk bør suppleres MED FDG-PET og / eller FP-CIT-SPECT som de er følsomme i å oppdage striatal involvering. I autosomale recessive epilepsier med indikative funn for en nevrodegenerativ sykdom, BØR vps13a enkeltgen test eller chorein Western blot brukes til å etablere riktig diagnose. Sistnevnte bør også vurderes i andre neurodegenerative lidelser uten genetisk definert etiologi.
Etikkerklæring
Skriftlig informert samtykke ble innhentet fra pasienten for deltakelse i studien. Skriftlig informert samtykke ble innhentet fra pasienten for publisering av denne saksrapporten.
Forfatterbidrag
JW, MR og SK deltok i pasientbehandling. JW skrev det første utkastet til manuskriptet. LF, HU og PM utarbeidet tall. Alle forfattere bidro til manuskriptrevisjon, leste og godkjente den innsendte versjonen.
Interessekonflikt
HU er aksjonær I Veobrain Gmbh, en spin-off Av University Medical Center Freiburg.
de resterende forfatterne erklærer at forskningen ble utført i fravær av kommersielle eller økonomiske forhold som kan tolkes som en potensiell interessekonflikt.
1. Walterfang M, Evans Et Leong Looi JESUS kristus, Jung TT, Danek En, Walker RH, et al. Neuropsykiatri av neuroacanthocytosis syndromer. Neurosci Biobehav Rev. (2011) 35: 1275-83. doi: 10.1016 / j.neubiorev.2011.01.001
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
2. Walker RH. Behandling av neuroacanthocytosis syndromer. Tremor Andre Hyperkinet. Mov. (2015) 5:346. doi: 10.7916 / D8W66K48
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
3. Jørgensen A, Jørgensen A, Jørgensen B, Jørgensen B, Jørgensen B, Jørgensen A, et al. Chorea-Akanthocytose. GeneReviews® Seattle, WA: Universitetet I Washington (1993).
4. Critchley EM, Clark DB, Wikler A. Akanthocytose Og Nevrologisk Lidelse uten Betalipoproteinemi. Arch Neurol. (1968) 18:134–40.
Pubmed Abstrakt
5. Levine IM, Estes JW, Looney JM. Arvelig nevrologisk sykdom med akanthocytose. Et nytt syndrom. Arch Neurol. (1968) 19:403–9.
PubMed Abstract | Google Scholar
6. Hardie RJ, Pullon HW, Harding AE, Owen JS, Pires M, Daniels GL, et al. Neuroacanthocytosis. en klinisk, hematologisk og patologisk studie av 19 tilfeller. Hjerne (1991) 114 (Pt 1A): 13-49.
PubMed Abstract | Google Scholar
7. Peluso S, Bilo L, Esposito M, Antenora A, Rosa ADR, Pappata S, et al. Chorea-akanthocytose uten chorea: utvidelse av den kliniske fenotypen. Parkinson Relatert Disord. (2017) 41:124–6. doi: 10.2016 / j. parkreldis.2017.05.013
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Scheid R, Bader B, Ott DV, Merkenschlager A, Danek A. Utvikling av mesial temporal lobe epilepsi i chorea-akanthocytose. Nevrologi (2009) 73: 1419-22. doi: 10.1212 / WNL.0b013e3181bd80d4
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
9. M. k., Kim SA, Grunseich C, Hefti MM, Crary JF, Danek A, et al. Hippocampal sklerose og mesial temporal lobe epilepsi i chorea-akanthocytose: et tilfelle med klinisk, patologisk og genetisk evaluering. Neuropathol Appl Neurobiol. (2017) 43:542–6. doi: 10.1111 / nan.12403
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert A, La Fougè C, Noachtar S, et al. Bilateral temporal lobe epilepsi bekreftet med intrakraniell EEG i chorea-akanthocytose. Beslag (2011) 20: 340-2. doi: 10.1016 / j.beslag.2010.12.007
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Marson AM, Bucciantini E, Gentile E, Geda C. Neuroacanthocytosis: kliniske, radiologiske og nevrofysiologiske funn i en italiensk familie. Neurol Sci. (2003) 24:188–9. doi: 10.1007 / s10072-003-0123-1
PubMed Abstrakt / Fulltekst / Google Scholar
12. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, et al. Choreinmangel fører til oppregulering av gefyrin og GABA(A) reseptor. Biochem Biophys Res Commun. (2006) 351:438–42. doi: 10.1016 / j.bbrc.2006.10.070
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Ehrlich DJ, Walker RH. Funksjonell neuroimaging og chorea: en systematisk gjennomgang. J Clin Mov Disord. (2017) 4:8. doi: 10.1186 / s40734-017-0056-0
PubMed Abstrakt / Fulltekst / Google Scholar
14. Connolly BS, Hazrati L-N, Lang AE. Neuropatologiske Funn i chorea-akanthocytose: ny innsikt i mekanismer som ligger til grunn For Parkinsonisme og anfall. Acta Neuropathol. (2014) 127:613–5. doi: 10.1007 / s00401-013-1241-3
PubMed Abstrakt / Fulltekst / Google Scholar
15. Sorrentino G, De Renzo A, Miniello S, Nori O, Bonavita V. Sen utseende av akanthocytter i løpet av chorea-akanthocytose. J Neurol Sci. (1999) 163:175–8.
PubMed Abstract | Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Choroo-akantocytose uten akantocytter. Revue Neurol. (2010) 166:100–3. doi: 10.1016 / j.neurol.2009.03.005
CrossRef Fulltekst | Google Scholar
17. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. Tap AV vps13c-funksjon i autosomal-recessiv Parkinsonisme forårsaker mitokondriell dysfunksjon og øker PINK1 / Parkin-avhengig mitofagi. Am J Hum Genet. (2016) 98:500–13. doi: 10.1016 / j.ajhg.2016.01.014
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M, et al. Recessive mutasjoner i >VPS13D forårsaker bevegelsesforstyrrelser i barndommen. Ann Neurol. (2018) 83: I6. doi: 10.1002 / ana.25204
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
19. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, et al. In vivo distribusjon og lokalisering av chorein. Biochem Biophys Res Commun. (2007) 353:431–5. doi: 10.1016 / j.bbrc.2006.12.059
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
20. Lang F, Pelzl L, Sch@ls L, Hermann A, Fö M, Sch Hryvffer TE, et al. Neuroner, erytrocytter og utover-de ulike funksjonene av chorein. Neurosignals (2017) 25: 117-26. doi: 10.1159/000485457
PubMed Abstrakt / Fulltekst / Google Scholar
21. Benner F, Afawi Z, Korczyn AD, Oliver KL, Pendziwiat M, Nakamura M, Et al. Beslag som presentere og fremtredende symptom i chorea-akanthocytose med c. 2343del VPS13A genmutasjon. Epilepsi (2016) 57: 549-56. doi: 10.1111 / epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar