Genetikken til leppe-og ganespalte
 Intro/abstractCleft leppe med eller uten ganespalte er en kompleks medfødt anomali som kan isoleres eller ses sammen med andre misdannelser. Det kan også være en del av fenotypen av et genetisk syndrom. Denne artikkelen fungerer som en gjennomgang av utbredelsen av leppe-og ganespalte, risiko for tilbakefall, og risiko for andre medfødte misdannelser. Genetiske syndromer og teratogene eksponeringer som er kjent for å være forbundet med orale spalter vil bli undersøkt. I tillegg vil genetiske tester ofte forespurt i pediatrisk klinisk genetikk innstilling for evaluering av pasienten med leppe-og ganespalte bli diskutert.
Intro/abstractCleft leppe med eller uten ganespalte er en kompleks medfødt anomali som kan isoleres eller ses sammen med andre misdannelser. Det kan også være en del av fenotypen av et genetisk syndrom. Denne artikkelen fungerer som en gjennomgang av utbredelsen av leppe-og ganespalte, risiko for tilbakefall, og risiko for andre medfødte misdannelser. Genetiske syndromer og teratogene eksponeringer som er kjent for å være forbundet med orale spalter vil bli undersøkt. I tillegg vil genetiske tester ofte forespurt i pediatrisk klinisk genetikk innstilling for evaluering av pasienten med leppe-og ganespalte bli diskutert.
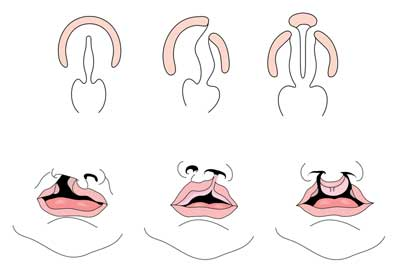
Leppe Med eller uten ganespalte (CL/CP) skiller seg fra en isolert ganespalte (CP) på embryonale, epidemiologiske og genetiske nivåer. Leppe-kløften skyldes vanligvis at den maksillære prominensen og den mediale nasale prominensen ikke smelter sammen mellom den femte og sjette uken med embryonisk utvikling. Normal ganeutvikling skyldes dannelsen av primær gane og sekundær gane. Den primære ganen dannes i uker seks til syv ved utvikling og fusjon av medial nasal, lateral nasal og maxillary prosesser. Den sekundære ganen stammer fra palatalhyllene (som utvikler seg fra de parrede maksillære prosessene i den første grenbuen) blir horisontal og fusjonerer, og danner de harde og myke ganene rundt den niende uken av embryonisk utvikling. Hyller smelter også sammen med den primære ganen og neseseptumet. (1)
muntlige kløfter er en av de vanligste fødselsdefektene sett i neonatal barnehage, med en samlet prevalens på 1.6 per tusen nyfødte over hele verden, MED CL/CP sett i omtrent en per tusen fødsler og CP sett i 0.6 per tusen fødsler. (2) DET er en høyere frekvens AV CL/CP hos personer Av Asiatisk, Afrikansk og Indiansk avstamning. CL / CP er også mer vanlig hos menn. DERIMOT er DET ingen signifikant forskjell i forekomst AV CP blant ulike etniske bakgrunner, OG CP er mer vanlig hos kvinner. (3) Risiko for tilbakefall i en familie avhenger av om spalten er isolert (uten andre kliniske funn tilstede) eller sett på som en del av et genetisk syndrom. De fleste tilfeller av muntlige spalter er isolert (ca 80%). Isolerte kløfter antas å ha multifaktoriell arv: de skyldes en kombinasjon av flere faktorer, både genetiske og miljømessige. Risikoen for tilbakefall (Tabell 1) øker når det er mer enn en berørt slektning. Risikoen for tilbakefall øker også jo mer alvorlig feilen er.

Leppe-Og ganespalte Kan ses ved andre medfødte misdannelser. Sannsynligheten for en genetisk eller teratogen etiologi øker de mer medfødte anomaliene som en pasient presenterer. Tilstedeværelse av andre problemer som intellektuell funksjonshemning, atferdsproblemer som autisme, dysmorfe egenskaper eller andre medisinske bekymringer vil også gjøre en genetisk lidelse eller en teratogen eksponering mer sannsynlig. Omtrent 13% av personer med leppe vil ha andre medisinske bekymringer eller anomalier. Antallet øker til 37% med leppe-og ganespalte og til 47% med ganespalte alene.
Prenatal eksponering for teratogene midler (som thalidomid, antikonvulsiva midler, alkohol, retinsyre og sigaretter) og maternal sykdom (som diabetes, rubella og folatmangel) har vist seg å øke risikoen for orale kløfter. Tilstedeværelsen av amniotiske bånd øker også risikoen for kløfter. Periconceptual folsyretilskudd er kjent for å redusere risikoen for orale kløfter.
Pierre Robin-sekvensen er en kraniofacial anomali preget av mandibulær hypoplasi eller mikrognati, sekundær u-formet ganespalte og glossoptose som fører til obstruktiv apnø og matvansker. Pierre Robin-sekvensen kan ses som en del av genetiske syndromer (22q11. 2 delesjonssyndrom, Sticklersyndrom; beskrevet nedenfor). (5)
det er hundrevis av genetiske syndromer forbundet med orale kløfter, inkludert cytogenetiske abnormiteter (aneuploidier, mikrodelesjoner) og enkeltgen (Mendelsk) lidelser. Bekreftelse av en genetisk diagnose er viktig for å bestemme prognose og etablere risiko for tilbakefall.
Aneuploidier Som Trisomi 13 og 18 har en sterk tilknytning TIL CL / CP. Trisomi 13 (AKA Patau syndrom) er assosiert med tre kopier av kromosom 13, eller ubalansert Robertsonian translokasjoner som involverer kromosom 13. Babyer født med denne tilstanden dør vanligvis i nyfødtperioden. Kliniske trekk inkluderer leppe-og ganespalte, vekstretardasjon, alvorlige misdannelser i sentralnervesystemet( inkludert holoprosencefali), mikrocefali, mikrotalmi, iriskolobom, fravær av øyne, misdannede ører, polydaktyly, knyttede never, vippebunn, medfødte hjertefeil og urogenitale defekter. Midtlinjeklyper (ellers svært sjeldne) kan ses ved trisomi 13 på grunn av risiko for midtlinjefeil, inkludert holoprosencefali. Trisomi 18 (AKA Edwards syndrom) skyldes vanligvis tre forskjellige kopier av kromosom 18, og er forbundet med dårlig postnatal utfall. Kliniske funksjoner inkluderer leppe-og ganespalte, utviklingshemming, manglende evne til å trives, medfødt hjertesykdom, hypertoni, mikrognati, kort sternum, lavt sett misdannede ører, knyttede hender, rocker bunn føtter, og hypoplastiske negler, blant andre. Trisomi 13 og 18 kan enkelt bekreftes eller utelukkes ved å gjøre kromosomanalyse (karyotyping).
Mikrodelesjonssyndromer involverer vanligvis sletting av en del av et kromosom. Disse slettingene kan være for små til å påvises ved standard karyotyping og kan kreve AT FISK (fluorescens in situ hybridisering) eller mikroarray-teknologi oppdages. Et velkjent mikrodelesjonssyndrom assosiert med spalt gane er 22q11. 2 delesjonssyndrom (aka Digeorge / Velocardiofacial syndrom). Palatale abnormiteter, inkludert velofaryngeal inkompetanse, submukosale kløfter, bifid drøvel og ganespalte, ses hos 69% av individer med 22q11.2 delesjon, og kan være En Del Av Pierre Robin-sekvensen. Andre kliniske funn inkluderer medfødt hjertesykdom, hørselstap, dysmorfe egenskaper, immunsvikt, hypokalsemi, nyreanomalier, matingsproblemer, skjelettavvik og psykiatriske lidelser. Omtrent 10% av tilfellene av 22q11.2 delesjonssyndrom antas å være familiære. Slettingen segregerer på en autosomal dominant måte.(6) Wolf-Hirschhorn syndrom, som skyldes en sletting i den korte armen av kromosom 4, er også forbundet med orale kløfter (hos 25% til 50% av berørte individer). Karakteristiske ansiktstrekk (inkludert fremtredende glabella fører til “gresk-kriger hjelm utseende”), medfødt hjertesykdom, utviklingshemming, beslag, unnlatelse av å trives, micrognathia, preauricular koder eller groper, og hypodontia kan også sees som en del av tilstanden.(7)
Enkeltgenlidelser med orale kløfter inkluderer Stickler syndrom, Treacher Collins syndrom og Van Der Woude syndrom, blant Mange andre. Stickler syndrom er en kollagenforstyrrelse med autosomal dominant og, mindre vanlig, autosomal recessiv arv. Vanlige trekk inkluderer ganespalte (sett som En Del Av Pierre Robin-sekvensen eller uten mikrognati), hørselstap( sensorinevral og ledende), skjelettfunn (tidlig oppstått artritt, spondyloepifyseal dysplasi), okulære anomalier (høy myopi, glasslegemeavvik) og karakteristiske ansiktsegenskaper (med underutvikling av kjeve og nesebro, midface retrusion). Genetisk testing For Stickler syndrom kan være komplisert, som mutasjoner i minst seks gener har blitt beskrevet i berørte individer. Omtrent 90% av pasientene Med Sticklers syndrom har mutasjoner I COL2A1-genet og har en autosomal dominant form av tilstanden.(8) Treacher Collins syndrom er en autosomal dominant tilstand preget av spalt gane med eller uten spalt leppe i 28% av berørte individer. Andre abnormiteter inkluderer hypoplasi av zygomatic bein og kjeven, ytre øret anomalier, coloboma av nedre øyelokk, ledende hørselstap, fravær av nedre øyevipper, preauricular hår forskyvning på kinnene, og choanal stenose eller atresi. Diagnosen Av Treacher Collins syndrom er basert på kliniske og radiografiske funn. Mutasjoner i minst tre gener er beskrevet, med mutasjoner I TCOF1 sett hos 78% til 93% av pasientene.(9) Van Der Woude syndrom er preget av tilstedeværelsen av medfødt, vanligvis bilateral, paramedian nedre leppe fistler (groper), eller noen ganger små hauger med en sinus kanalen som fører fra en slimete kjertel av leppen, og muntlige spalter (inkludert CL/CP og CP). Van Der Woude er en autosomal dominant tilstand assosiert med mutasjoner I IRF6-genet (10). Testing for single-gen eller multi-gen forhold krever direkte analyse av genet ved sekvensering og/eller sletting / duplisering analyse (FOR EKSEMPEL MLPA).
Gitt at genetiske syndromer med leppe-og ganespalte kan være forbundet med aneuploidier, kromosommikrodeleksjoner/mikroduplikasjoner eller enkeltgenforstyrrelser, kan genetisk testing være en komplisert prosess. En grundig medisinsk historie, en tre-generasjons stamtavle, en graviditet historie, og en dysmorfologi eksamen av en klinisk genetiker kan avklare det kliniske bildet og tillate målrettet genetisk testing. Nyere teknologier, inkludert microarray, vil tillate identifisering av små mikrodeletjoner og mikroduplikasjoner som tidligere ble savnet av standard karyotyping. Dessverre fører denne teknikken også til identifisering av slettinger og duplikasjoner av ukjent klinisk betydning, og kompliserer den genetiske rådgivningsprosessen. Testing for single-gen lidelser eller Mendelian lidelser krever klinisk tilgjengelighet av genetisk testing for ønsket gen. Det kan også være dyrt hvis det ikke dekkes av medisinsk forsikring. Nye teknologier som Neste Generasjons sekvensering, eksomsekvensering eller genomsekvensering (kjent kollektivt som genomiske tester) har nå blitt klinisk tilgjengelige. Ved å analysere hundrevis til tusenvis av gener samtidig, øker disse testene signifikant diagnostisk kraft og utbytte. Sammenlignet med andre teknikker, kan disse testene gi et svar raskere og på en mer kostnadseffektiv måte. I forskningsfeltet har eksom-og genomsekvensering ført til identifisering av nye gener, samt utvidelse av kliniske egenskaper og spektrum for genetiske mutasjoner. Som med microarray-teknologi kan genomiske tester oppdage syndromer som ikke er relatert til pasientens presentasjon og / eller grunn til testing. Gitt den iboende kompleksiteten av genetisk testing, informert samtykke er nødvendig.
Konklusjon
selv om leppe-og ganespalte er en isolert anomali i de fleste tilfeller, er det en sterk sammenheng mellom orale kløfter og andre anomalier og genetiske syndromer. En genetisk evaluering av en klinisk genetiker og en genetisk rådgiver er avgjørende for å forutse veiledning og å bestemme risiko for tilbakefall. Genetisk testing, som krever informert samtykke, kan koordineres og tolkes under en genetisk evaluering.
Anya Revah, MS, er senior genetisk rådgiver Ved Divisjon For Medisinsk Genetikk Ved Maimonides Spedbarn Og Barnesykehus I Brooklyn, New York. Hun er også et aktivt medlem Av Maimonides Medical Center Og Kings County Hospital Cleft Leppe og Gane Tverrfaglig Team. Hun har En Mastergrad I Vitenskap I Genetisk Rådgivning fra Boston University I Boston, Massachusetts.
1. Sadler TW. Langmans Medisinske Embryologi. Niende Utgave. Sider 390-395.
2. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. For Det Nasjonale Fødselsdefektforebyggende Nettverket. Oppdaterte nasjonale fødselsprevalensestimater for utvalgte fødselsskader i Usa. 2004-2006. Fødselsdefekter Forskning (Del A): Klinisk Og Molekylær Teratologi 2010; 88:1008-1016.
3. Fraser FC. Genetikken til leppe-og ganespalte. Er. J. Hum. Genet. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC, et al. Periconceptual folatinntak ved supplement og matinntak reduserer risikoen for ikke-syndromisk spalt leppe med eller uten spalt gane. Forrige Med 2004; 39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Utviklings-og genetiske perspektiver På Pierre Robin-sekvensen. Er. J. Med. Genet. 2013; 163C:295-305.
6. McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11. 2 Sletting Syndrom. September. 23, 1999. . I: Pagon RA, Adam MP, Ardinger HH, et al., redigeringsprogram. GeneReviews . Seattle (WA): Universitetet I Washington, Seattle; 1993-2014. Tilgjengelig fra: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Jc, Jc, Jc, Et al. Wolf-Hirschhorn Syndrom. Apr. 29, 2002. . I: Pagon RA, Adam MP, Ardinger HH, et al., redigeringsprogram. GeneReviews . Seattle (WA): Universitetet I Washington, Seattle; 1993-2014. Tilgjengelig fra: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, Ala-Kokko L. Sticklers Syndrom. Jun. 9, 2000. . I: Pagon RA, Adam MP, Ardinger HH, et al., redigeringsprogram. GeneReviews . Seattle (WA): Universitetet I Washington, Seattle; 1993-2014. Tilgjengelig fra: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs EW. Treacher Collins Syndrom. Jul. 20, 2004. . I: Pagon RA, Adam MP, Ardinger HH, et al., redigeringsprogram. GeneReviews . Seattle (WA): Universitetet I Washington, Seattle; 1993-2014. Tilgjengelig fra: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy S, et al. IRF6 – Relaterte Lidelser. Oktober. 30, 2003. . I: Pagon RA, Adam MP, Ardinger HH, et al., redigeringsprogram. GeneReviews . Seattle (WA): Universitetet I Washington, Seattle; 1993-2014. Tilgjengelig fra: http://www.ncbi.nlm.nih.gov/books/NBK1407/.