Grenser I Pediatri
Introduksjon
Familiære eller arvelige hjertearytmier omfatter betydelige prosenter av arytmier Og også årsak til plutselig hjertedød (SCD) (1, 2). I løpet av de siste to tiårene har forskere og klinikere gitt enorm innsats for å løse de intrikate og komplekse mekanismene for medfødte familiære arytmier (1-8). For å forstå mekanismen for arytmogenese, må vi vite grunnleggende om hjertecellulær struktur og deres elektrofysiologiske egenskaper. Hjertemyocytter er de viktigste fungerende cellene i hjertet, og de er i stor grad koblet slik at impulser forplanter seg raskt og jevnt. Kardiomyocytter er skilt fra hverandre av en spesialisert grense kalt den interkalerte disken; gapkrysseproteiner, hjerte desmosomer og ionkanaler er plassert i den interkalerte disken (9). Gapkryssene består av tettpakkede forbindelser som tillater intercellulær utveksling av små molekyler og tillater også å strømme eksitatoriske strømmer fra en celle til sin nabocelle. Desmosomer sammen med adherens-kryssene er ansvarlige for de mekaniske vedleggene til individuelle kardiomyocytter. Alle disse komponentene i de interkalerte diskene er sekvensielt segregerte og hver komponent utøver sin unike funksjon; forstyrrelse av den ene komponenten påvirker funksjonen til andre komponenter, som predisponerer hjertet for å utvikle arytmier (9-11). Hjerte ionekanaler er poreformende proteinkomplekser som gir spenning gated og intrikat koordinert innover og utover bevegelse av ioniske strømmer over cellemembranene, avgjørende for hjerterytmegenerering og forplantning. Lang QT-syndrom (lqts), kort QT-syndrom (sqts), syk sinus syndrom (sss), hjerteledningsdefekt (CCD), BrS, katekolaminerg polymorf ventrikulær takykardi (CPVT), tidlig repolariseringssyndrom (ERS) og familiær Atrieflimmer (AF) er de for tiden kjente hjertekanalopatier, som kan oppstå på grunn av en enkelt eller flere feil i generene knyttet til hjerterytmegenerering og forplantning.
i denne gjennomgangen vil vi beskrive utelukkende PÅ lqts knyttet arytmier, dets patofysiologi og tiden tilgjengelig klinisk ledelse. Vi har pionerer i å belyse genetisk patologi i familiære hjertearytmier I Saudi-Arabia (1, 12-14), vi utfører fortsatt kardiogenetiske undersøkelser i Saudi-Arabia. Vi vil også legge til de nylig publiserte dataene OM LQTS fra et team På King Faisal Specialist Hospital and Research Centre, Riyadh (15).
LANG QT-Syndrom
Medfødt Lqts er en arvelig lidelse definert ved forlengelse AV QT-intervallet på elektrokardiogram (EKG). Pasienter med alle former FOR LQTS er predisponert for ventrikulær takyarytmi, torsades de pointes (TdP) som fører til tilbakevendende synkope eller SCD. I mange tilfeller kan synkope eller plutselig død være den første og eneste manifestasjonen. LQTS påvirker anslagsvis 1 av 2000 personer over hele verden (16). Kjennetegnet FOR LQTS er forlengelse AV QT-intervallet PÅ EKG (korrigert for hjertefrekvens, dvs. QTc). Normale verdier Av QTc er 440 ms hos menn og 450 ms hos kvinner. Hos barn er alder og kjønnsavhengige verdier relevante. Nylig konsensus anbefaling FOR lqts diagnose er følgende (17, 18):
1. Lqts er diagnostisert:
a. i nærvær AV en lqts-risikoscore var det 3,5 i fravær av en sekundær årsak TIL qt-forlengelse og / eller
b. i nærvær av en utvetydig patogen mutasjon i ET av lqts-genene eller
c. VED qt-intervall korrigert for hjertefrekvens ved Bruk Av Bazetts formel (QTc) ≥500 ms ved gjentatt 12-leders elektrokardiogram og ved fravær av en sekundær ÅRSAK til qt-forlengelse.
2. LQTS kan diagnostiseres i Nærvær Av QTc mellom 480 og 499 ms i gjentatte 12-bly Ekg hos en pasient med uforklarlig synkope i fravær av en sekundær årsak TIL qt-forlengelse og i fravær av en patogen mutasjon.
Pasienter med qtc-varighet ≥500 ms hadde en signifikant høyere kumulativ sannsynlighet for å oppleve sitt første synkope sammenlignet med pasienter med QTc-varighet <500 ms fra fødsel til alder 20 år (19). Men pasienter som har opplevd 2, 3 og 4 synkope episoder, var risikoen for en påfølgende synkope episode nesten identisk mellom pasienter som hadde smal Eller langvarig QTc varighet (19). DET molekylære grunnlaget FOR LQTS er heterogent og til dags dato har mutasjoner i 13 forskjellige gener blitt beskrevet kausale FOR LQTS (1-8, 19, 20). En genetisk defekt finnes vanligvis hos 70% AV LQTS-pasientene i ett av disse 13 genene (1-8, 19, 20). Blant de for tiden kjente 13 forskjellige typer LQTS, er DE vanligste LQTS1, LQTS2 og LQTS3, på grunn av defekter i hjerte ionkanalgener, HENHOLDSVIS kcnq1, KCNH2 og SCN5A. Nitti prosent av ALLE LQTS kausale mutasjoner finnes i disse tre genene (1-8, 19, 20). Mutasjoner i de resterende 10 genene er sjeldne og utgjør bare ∼10% av alle DE kjente LQTS-mutasjonene.
Lang QT-syndrom er vanligvis en autosomal dominant sykdom, men noen ganger kan flere mutasjoner i et enkelt gen eller i forskjellige gener bli funnet hos 5-10% av pasientene MED LQTS (21, 22). Pasienter med flere mutasjoner kan vise en lengre QTc sammenlignet med de med en enkelt mutasjon, og slike pasienter har også en 3,5 ganger økt risiko for livstruende hjertehendelser (21, 22)
LQTS1
Mutasjoner I kcnq1-genet er den vanligste formen for ALLE LQTS og referert TIL SOM lqts type 1 (LQTS1). Femti prosent av ALLE LQTS-kausale mutasjoner finnes I kcnq1-genet (20). KvLQT1 (Også Kalt Kv7.1) er et protein laget AV kcnq1-genet, og proteinet danner tetramerer i endoplasmatisk retikulum inne i cellene, som formentlig sammonterer med proteinmink (kodet AV KCNE1) og transporteres deretter til plasmamembranen i hjertemyocytene, hvor de formidler en sakte aktiverende strøm som akselererer repolariseringen av handlingspotensialet i hjertevev, denne strømmen er kjent som IKs (Figur 1). LQTS1 kausale kcnq1-mutasjoner er for det meste missense-mutasjoner, og i sjeldne tilfeller kan de være rammeskiftmutasjoner I Den C-terminale regionen (23). Hjertearytmi hos kcnq1-mutasjonsbærere utløses av adrenerge drifter, f.eks. emosjonelt stress, fysisk anstrengelse, dykking, svømming (24-26). Pasienter med mutasjoner på transmembrane domener Av KvLQT1 har høyere risiko FOR LQTS-relaterte hjertehendelser og har større følsomhet for sympatisk stimulering (26, 27). Pasienter med missense-mutasjoner har økt risiko sammenlignet med pasienter med ikke-sansende eller avkortende mutasjoner (27, 28). Variasjon i 3 ‘ – UTR i kcnq1-genet påvirker også arytmiefølsomheten betydelig, antagelig ved å påvirke uttrykket av genet (29). Pasienter med arytmier på grunn AV kcnq1-mutasjoner reagerer ganske godt på β-blokkere, men noen pasienter kan fortsatt være mindre responsive eller til og med resistente mot denne medisinen. I en fersk artikkel, Barsheshet et al. (30) hevdet at pasienter med mutasjoner utenfor cytoplasmisk loop (c-loop) – regionen I KvLQT1 er mindre mottakelige for β-blokkere.
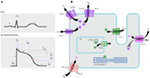
Figur 1. Ioniske strømmer som bidrar til det ventrikulære virkningspotensialet (A) og skjematisk fremstilling av en kardiomyocyt som viser (bare) de proteinene som er involvert i patogenesen av arvelige arytmisyndrom (B). I (A) er handlingspotensialet justert med sin omtrentlige virkningstid under EKG. I (B) er ankyrin-B, et adapterprotein involvert i lang QT-syndrom type 4, ikke avbildet.
Homozygote eller sammensatte heterozygote mutasjoner I kcnq1-genet, som kan forårsake den recessive formen av sykdommen, Jervell og Lange-Nielsen syndrom (JLNS), type 1 (31), er ganske sjeldne. JLNS-pasienter lider også av alvorlige hjertearytmier og døvhet (31, 32). Pasienter med kcnq1-mutasjoner som forårsaker JLNS har vanligvis ingen funksjonell IKs (28, 31-33). Det er mulig at noen pasienter ikke kan ha døvhet til tross for homozygote eller sammensatte heterozygote mutasjoner I KCNQ1, slike tilfeller omtales som autosomal recessiv LQTS1 (13, 32). Hos disse pasientene kan liten mengde funksjonell IKs-strøm (<10% av Total IKs) fortsatt være tilstede, noe som opprettholder hørselsfunksjonen, men deres hjerterytmefeil er like alvorlige som HOS JLNS-pasienter (13, 32).
LQTS2
Denne typen LQTS er like utbredt som LQTS1, og utgjør 35-40% AV lqts-pasientene med en påviselig mutasjon (1, 20, 24, 25). KCNH2-koder FOR herg-proteinet (Kv11.1), som er α-underenheten til Den raskt aktiverende forsinkede likriktaren K+ current (IKr). Patogene mutasjoner i dette genet som reduserer Kv11. 1 kanalfunksjonen forlenger varigheten AV QT-intervallet (Figur 1), og er kausale TIL LQTS2. Tjuefem prosent av synkopale angrep i LQTS2 oppstår under hvile/søvn, og bare 13% av synkopale angrep ble rapportert å forekomme under trening (25, 34). Plutselige oppsiktsvekkende lyder, f.eks. alarmklokker, dørklokker, telefonringer, utløser vanligvis synkopale angrep hos disse pasientene (24, 34). Pasienter med mutasjoner I poredannelsesområdet I Kv11.1 (kodet AV kcnh2-genet) er utsatt for høy risiko for arytmirelaterte hjertehendelser sammenlignet med pasienter med ikke-poreregionsmutasjoner (35). Hos nyfødte er 2: 1 Atrioventrikulær (Av) blokk fortrinnsvis assosiert MED kcnh2-mutasjoner (36). Komplett av-blokk komplisert AV LQTS ble også funnet hos 17% av voksne pasienter med en mutasjon I kcnh2 genet (37). Homozygote mutasjoner I KCNH2 er sjeldne, og når de er tilstede, lider pasienter av en alvorlig form FOR LQTS, med 2:1 av-blokk og alvorlige ventrikulære arytmier, under intrauterin stadier samt etter fødselen (11, 38-40). I tillegg til de mange mutasjonene som er beskrevet I lqts2-patologi, kan vanlige polymorfe varianter I kcnh2-genet også modulere sykdommens alvorlighetsgrad. Et spennende eksempel ER k897t polymorfisme (SNP) I KCNH2, som er tilstede i 33% av befolkningen generelt (41). K897T ble rapportert å forverre patogeniteten TIL kcnh2-mutasjonene (42, 43).
LQTS3
Nav1.5 er den poreformende α-underenheten til den spenningsavhengige hjerte Na+ – kanalen, det er et integrert membranprotein kodet AV scn5a-genet og er involvert i initiering og ledning av hjerteaksjonspotensialer. Kardiale Na+ – kanaler består av en poreformende α-underenhet (kodet AV SCN5A) og en eller flere tilleggsenheter.
LQTS3 skyldes gevinst på funksjonsmutasjoner som forstyrrer rask inaktivering av α-subenheten (Figur 1), og en mutasjon I SCN5A-genet er beskrevet hos <10% AV ALLE LQTS-pasienter med en mutasjon (1, 20, 24). LQTS3-pasienter opplevde majoriteten (39%) av hjertehendelsene under søvn/hvile (25), og om lag 13% av hendelsene ble rapportert å forekomme under trening (25). I flere tilfeller ble en ENKELT SCN5A-mutasjon vist å utøve to eller til og med tre forskjellige fenotyper av arytmier i samme familie, for EKSEMPEL LQTS, BrS eller CCD (44-47). Mannlige pasienter med lqts3-mutasjon kan utvikle symptomer mye tidligere enn kvinnelige pasienter (48). Både heterozygote OG homozygote SCN5A-mutasjoner er beskrevet I LQTS3 med funksjonell 2: 1 av-blokk (49). Vi har nylig funnet En Iransk familie med 1507_1509delqkp-mutasjonen i flere familiemedlemmer, hvor pasientene hadde kombinert LQTS og CCD (data ikke vist). Denne mutasjonen har blitt rapportert hos pasienter fra andre land, noe som tyder på at dette er en tilbakevendende og hot spot mutasjon . Mutasjoner med slike tap – og gevinst-av-funksjon egenskaper i ulike faser av aksjonspotensialet ble også rapportert i 1493delk og 1795insd mutasjoner (44, 51).
Noen GANGER kan EN SNP også utøve patogen effekt på bærerne. S1103Y er en vanlig variant I scn5a-genet, tilstede hos 13% Av Afroamerikanerne (52). Bærere med denne varianten har økt risiko for arytmier og krybbedød (SIDS) (52).
LQTS4
lqts4 representerer den første ikke-kanalformen AV LQTS. En mutasjon I ANK2, et adapterprotein, fører til intracellulær kalsiumoverbelastning som bidrar TIL LQTS4 (53, 54). I TILLEGG TIL qt-forlengelse kan pasienter med dette syndromet ha sinus bradykardi, paroksysmal AF og CPVT (54). DEN patogene effekten AV ANK2-mutasjonene kan være moderat til alvorlig, og de kliniske uttrykkene avhenger av alvorlighetsgraden av mutasjonen.
LQTS5
Mutasjoner I KCNE1 er assosiert MED LQTS5 (Figur 1) (55, 56). Heterozygote kcne1-mutasjoner reduserer IKs ved å utøve en dominerende negativ effekt på det medfølgende normale allelet, og fører til forsinket kardial repolarisering (Figur 1), som er ansvarlig for økt risiko for arytmier (6). Pasienter med homozygote kcne1-mutasjoner lider AV JLNS (type 2) (57, 58).
D85N er en polymorfisme I kcne1-genet, tilstede i 0.7-1% av befolkningen generelt (41). I en studie Av Nishio et al. (59), d85n polymorfisme ble hyppigere funnet HOS LQTS-pasientene, noe som gjør det til en risikogenotype i LQTS-patologi (potensielt bare I Den Asiatiske befolkningen). I Europa ble D85N rapportert hos 5% av oppkjøpte LQTS (aLQTS) pasienter (i en kohort på 32 pasienter), som hadde opplevd TdP (60).
LQTS6
kcne2-genkoder For MinK-relatert peptid 1 (MiRP1), en formodentlig β-underenhet Av Hjertets kaliumkanal IKr (Figur 1). Mutasjoner I kcne2-genet kan også føre til defekter i den raskt aktiverende komponenten Av Forsinket likeretterkaliumstrøm (IKr), patologisk grunnlag FOR LQTS6 (61). Auditiv / akustisk stimulus som vekkerklokke støy, ringeklokke etc. kan provosere synkopale angrep i kcne2-mutasjonsbærere, lik KCNH2-mutasjon (62).
LQTS7
Dette syndromet er også kjent Som Andersen–Tawil syndrom (Ats). ATS er en sjelden lidelse, manifestert av sporadisk synkope og hjertestans. EKG-funksjoner inkluderer mild qt-intervall forlengelse, unormale u-bølger, hyppig ventrikulær ektopi, toveis ventrikulær takykardi (VT) og polymorf VT. Dette syndromet viser også ekstrakardiale egenskaper, for eksempel skjelettmuskel periodisk lammelse og utviklingsproblemer, som spalt gane, lavt sett ører, kort statur og utviklingsfeil i lemmer (63). Flertallet av klinisk diagnostiserte ATS-pasienter ble rapportert å ha en mutasjon I KCNJ2 (63). KCNJ2 koder for en poreformende underenhet av innvendig korrigerende kaliumkanaler (IK1) (Figur 1) (64, 65).
LQTS8
også Kjent Som Timothy syndrom (TS), pasienter viser alvorlig qt-forlengelse på Ekg, som er kombinert med syndaktyly, skallethet ved fødselen, og små tenner i 100% av tilfellene og mindre penetrant hjerte strukturelle misdannelser, mental retardasjon, autisme, og ansikts dysmorfe funksjoner (66). DET er to undertyper: ts1 (klassisk) OG TS2 (sjelden form).
TS2 er kardiologisk mer alvorlig ENN TS1 (66, 67). TS2-pasienter mangler også syndaktyly (67). Mutasjoner i α – 1-underenheten av L-type kalsiumstrøm (ica-L) som koder for genet CACNA1C fører til BEGGE former FOR TS (LQTS8). Det er alternativt spleiset, gjensidig utelukkende exon-8 i cacna1c-genet, for klarhet er de navngitt som exon-8 og exon-8a. i hjerte og hjerne, hvor CACNA1C hovedsakelig uttrykkes, er exon-8 funnet i ∼80% av mRNA-transkripsjonene og exon-8a er til stede ∼20% transkripsjoner (66). G406R mutasjon i exon – 8A er årsak til klassisk FORM AV TS (TS1) og G402S i exon-8 ble rapportert i severer I TS2. Et nytt tillegg til denne listen er ala1473gly mutasjon, som er beskrevet i ET TS-spedbarn med ytterligere utvidet fenotype (68). Alle mutasjoner var enten de novo eller mosaic i moderen, og alle resultatgevinster av funksjonen Til ica-l-kanalen (66-69).
LQTS9 og LQTS10
Mutasjoner I CAV3 eller SCN4B gir gevinst av funksjon sent I INa, noe som forårsaker EN LQTS3-lignende fenotype (70-74). De er KJENT SOM LQTS9 (assosiert MED CAV3 mutasjon) OG LQTS10 (assosiert MED SCN4B mutasjon).
Caveolae er pent beskrevet Av Engelman et al. (72) som “små grotter” i plasmamembranen. De er små ubelagte groper og regnes som stedet for viktige dynamiske og regulatoriske hendelser ved plasmamembranen (72, 73). Caveoliner er de viktigste proteinene i caveolae, og caveolin-3 (kodet av GENET CAV3) er spesielt funnet i kardiomyocytter og skjelettmuskelceller. Flere hjerte ionekanaler er spesielt rapportert å være lokalisert i caveolae ekstrahert fra hjerte myocytter som er beriket i caveolin-3 (72, 73). I tillegg er komponenter i den adrenerge reseptorsignalkaskaden også til stede i caveolae-berikede membraner (72, 73).
SCN4B koder For NaVß4, som er en ekstra β-underenhet av hjertenatriumkanalen. Så langt har bare EN mutasjon (L179F) blitt rapportert i dette genet i En Meksikansk familie med flere berørte familiemedlemmer (74). Mutasjon i dette genet ble funnet å resultere i gevinst av funksjon Av Nav1 .5 strøm (74).
LQTS11
i hjertet medieres sympatisk regulering av hjertets virkningspotensial varighet (APD) ved aktivering av β-adrenerg reseptor (β-AR), som krever sammenstilling AV AKAP9 (Yotiao) MED α-underenheten (KvLQT1) i IKs-kanalen. Mutasjon I AKAP9 forårsaker LQTS11 (75). Hittil er det kun rapportert en mutasjon, S1570L, I AKAP9 (75).
LQTS12
Mutasjon i α-1-Syntrofin (SNTN1) genet er årsak TIL LQTS12. Mutasjon i dette genet fører til gevinst av funksjon av hjertenatriumkanalen (Nav1.5), som er det patologiske grunnlaget FOR LQTS12 (76).
LQTS13
g proteinkoblet, innadrettet kaliumkanalunderenhet (Kir3.4) er kodet AV kcnj5-genet. En funksjonstapsmutasjon i dette genet kan forårsake LQTS13 (77). Hittil har BARE EN mutasjon, G387R, blitt beskrevet i En Kinesisk familie med ni pasienter som har denne mutasjonen. Redusert plasmamembranuttrykk Av Kir3. 4 ble foreslått som patologi AV LQTS hos pasientene.
Ervervet LQTS
i tillegg til medfødt lqts finnes også en annen variant av LQTS kjent som aLQTS, som skyldes faktorer og stoffer som reduserer kaliumflux og svekker myokardets evne til å repolarisere. Anerkjente forhold er kvinnelig kjønn, hypokalemi og legemidler som hemmer hjertekaliumkanaler (78-80). En rekke vanlige foreskrevne legemidler kan også fortrinnsvis binde OG blokkere herg-kanalen (Kv11. 1, et protein kodet AV kcnh2-genet) og predisponerer for aLQTS (79, 80). Nylig ble det vist at blokkering av IKs også kan bidra til legemiddelindusert alqt, spesielt når repolariseringsreserven er kompromittert (81). Fluoksetin og norfluoksetin ble funnet å undertrykke IKs-egenskapene, både in vivo og in vitro, og førte til merket LQTS (81). Polymorfismer, D85N i mink (gene KCNE1), T8A, Q9E I MiRP1 (gene KCNE2), som er antatt å være underenheter av iks-og IKr-kanalene, ble rapportert å forårsake aLQTS (82, 83). Autoimmune LQTS er også rapportert hos en pasient med IgG som inneholder anti-HERG antistoffer (84).
Elektrokardiografiske Trekk ved De Tre Vanlige Formene For LANG QT-Syndrom
Typiske st-t-bølgemønstre finnes hos de fleste GENOTYPEDE LQTS-pasienter og kan brukes til å identifisere LQTS1, LQTS2 og muligens lqts3-genotyper (85, 86).
lqts1-formen av LQTS er forbundet med en bred T-bølge uten forkortelse AV QT-intervallet ved trening (Figur 2a). LQTS2 er forbundet med lav amplitude, ofte bifid, T-bølger (Figur 2b). LQTS3 er forbundet med et langt iso-elektrisk segment og en smalbasert, høy T-bølge (Figur 2c). Pauseavhengighet av TdP-debut ved medfødt LQTS er genotypespesifikk, dominerende I LQTS2, men nesten fraværende i LQTS1 (87).

Figur 2. Elektrokardiogramopptak fra pasienter MED LQTS1, LQTS2 og LQTS3. (A) 12-bly EKG av en 18 år gammel mann med en kcnq1-mutasjon. QT-intervallet er forlenget (QTc = ±500 ms). St-segmentet har en bred base og relativt stor amplitude. Ledningsintervallet er normalt (standardkalibrering). (B) 12-bly EKG av en 14 år gammel jente MED EN KCNH2-mutasjon. QT-intervallet er forlenget(QTc ± 520 ms). St-segmentet er hakket i bly V3 og har relativt lav amplitude i ekstremitetsledningene. Ledningsintervallet er normalt (standardkalibrering). (C) 12-bly EKG av en 12 år gammel gutt MED EN SCN5A mutasjon. QT-intervallet er forlenget(qtc ± 600 ms). ST-segmentet har et langt (nesten) iso-elektrisk segment med en stor, skarp Og smal T-bølge. Ledningsintervallet er normalt (standardkalibrering).
selv om mønstre kan foreslå en spesifikk genotype AV LQTS, har unntak ofte blitt funnet.
Genotype-Fenotype
Lang QT-syndrom er en autosomal dominant sykdom. Genotype-og fenotypeanalyse blant heterozygote mutasjonsbærere har blitt utført ganske omfattende hos LQTS1 -, LQTS2-og LQTS3-pasientene. I barndommen var risikoen for hjertehendelser signifikant høyere hos lqts1-menn enn hos lqts1-kvinner, mens det ikke ble observert signifikante kjønnsrelaterte forskjeller i risiko for hjertehendelser blant lqts2-og LQTS3-pasienter (88-91). I voksen alder (også etter 40 år) kunne lqts1 og lqts2 kvinner ha signifikant høyere risiko for hjertehendelser enn respektive menn (88-91). Generelt synes dødeligheten av hjertehendelser å være dominerende hos LQTS3-pasienter enn HOS lqts1-og LQTS2-pasienter (91). Kvinner med LQTS har redusert risiko for hjertehendelser under graviditet, men risikoen øker ganske i 9-måneders postpartumperioden, spesielt hos kvinner med mutasjon I kcnh2-genet (92).
Plutselig hjertedød hos barn kan også være forårsaket av mutasjoner i hjerte ion kanal gener (93-96). Omtrent 28% av barna med uforklarlig SCD (mellom 1 og 18 år, gjennomsnittsalder: 12,3 ± 3,8 år) var bærere AV mutasjoner I LQTS – kausale gener (97). I SIDS virket mutasjoner I SCN5A dominerende (98, 99), men mutasjoner i KCNQ1, KCNH2, KCNE2 OG CAV3, SCN4B og SCN3B ble også funnet (100, 101). Intrauterin føtal dødsfall ble også rapportert på grunn av defekter i hjerte ionkanalgener (12, 102).
Klinisk Behandling av LQTS
Opphør av alle legemidler som er kjent for å forlenge QT-intervallet og også korrigering av elektrolyttforstyrrelser og/eller utløsende metabolske tilstander bør være hovedfokus ved behandling AV (ervervede) lqts-pasienter. SYMPTOMER på LQTS er ofte adrenergisk mediert, derfor anbefales det generelt å begrense pasientens deltakelse i atletiske aktiviteter (24, 25). Bærebjelken i klinisk behandling for lqts er β-blokade. Langtidsvirkende preparater av propranolol, nadolol og metoprolol brukes vanligvis, og effekten av dem ved β-blokkering vurderes ved blunting av treningspuls (f. eks. ved >20%) (24, 25). Blant alle β anses propranolol og nadolol å være bedre enn metoprolol hos symptomatiske pasienter (103). Videre kan β-blokade også brukes som profylaktisk behandling hos lydløse mutasjonsbærere for å redusere SCD (24). I en studie Av Barsheshet et al. (30) pasienter med c-loop missense-mutasjoner i kcnq1-genet hadde høy risiko for livstruende hjertehendelser, og de hadde betydelige fordeler ved behandling med β. Da kvinner med LQTS2 har økt risiko i løpet av 9-måneders postpartumperioden, bør β foreskrives for å redusere hjertehendelser i løpet av denne høyrisikoperioden (92). Implanterbare kardioverterdefibrillatorer (Icd) kan vurderes hos pasienter med tilbakevendende synkope til tross for β-blokkerbehandling eller hos pasienter med høy risiko for hjertestans (f. eks. symptomatisk lqts2 og LQTS3 med dokumentert qtc-forlengelse). Venstre kardial sympatisk denervering (lcsd) anbefales for HØYRISIKO LQTS-pasienter der EN ICD er kontraindisert eller nektet og β-blokkere enten ikke er effektive, ikke tolereres, ikke aksepteres eller kontraindisert (18, 104). Jlns-pasienter har Vanligvis En QTc >500 ms og har også høy risiko, β-blokkere har utilstrekkelig effekt hos slike pasienter hvor tidlig BEHANDLING MED ICD ble anbefalt (18, 31).
LQTS: Saudi Perspective
Første rapport OM LQTS Fra Saudi-Arabia ble publisert i 1993 Fra Riyadh Armed Forces Hospital (105). Fire spedbarn og små barn mellom 6 og 48 måneder med tidligere tilbakevendende anfall, fra en enkelt familie, ble diagnostisert SOM LQTS (105). Familiehistorie viste at to andre utvidede familiemedlemmer hadde lignende episoder av plutselig bevissthetstap og tre familiemedlemmer døde plutselig (105). I alle tilfeller var første diagnose epilepsi (105). Flere år senere er det rapportert to sporadiske kasusrapporter med relativt skarpere variant av neonatal LQT kombinert med 2:1 av-blokk (106, 107). Alle disse publiserte kliniske rapportene var uten noen genetiske funn som kunne forklare patofysiologien TIL LQTS i dem (105-107).
vi har for første gang rapportert genetiske defekter som patologisk grunnlag for LQTS hos lignende Pasienter Fra Saudi-Arabia. Vi har undersøkt Seks-Saudiske familier med synkope og plutselige uforklarlige dødsfall av foster, nyfødte og barn (12-14). Autosomal recessiv LQTS1 ble diagnostisert hos barn fra to familier(Figur 3). Autosomal recessiv LQTS2 ble diagnostisert i to familier(Figur 4). I en familie ble en kvinnelig pasient diagnostisert med autosomal dominant LQTS2 (Figur 5), pasienten hadde synkopale angrep under postpartum gjenopprettingstid på sykehuset, noe som er svært vanlig hos kvinner MED kcnh2 mutasjonsbærer. Hos alle våre pasienter førte identifisering Av patogene mutasjoner i LQTS kausale hjerte ionkanalgener til en bekreftet klinisk diagnose, som ble feildiagnostisert som epileptiske kramper Før de ble henvist til oss (13, 14) Nylig, Shinwari et al. (15) Fra King Faisal Specialist Hospital and Research Centre rapporterte EN LQTS1 kausal kcnq1 mutasjon, H258P, i en stor familie med 12 berørte individer. Bare to bærere var symptomatiske, Deres QTc var > 500 ms, og β-blokkere undertrykte de kliniske symptomene hos en pasient og den andre symptomatiske pasienten krevde EN ICD.
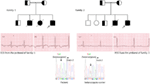
Figur 3. Stamtavle tegning av familien-1 og 2 med autosomal recessiv LQTS1, probands er vist med en pil. Ekg fra probands i to familier er vist i midten, med en QTc på henholdsvis 557 og 529 ms. Intronisk mutasjon, c.387 -5 T > A i kcnq1-genet ble funnet hos pasientene (vist nederst). Mutasjon ble vist med en pil. Ikke-fylte sirkler og firkanter er ikke-bærere for mutasjonen. Berørte personer vises som fylte sirkler (kvinner) og firkanter (menn). Halvfylte firkanter og sirkler er individer med heterozygot mutasjon. Avdøde personer er angitt med skråstreker, probands er angitt med en pil og consanguineous ekteskap er angitt med = Exon-intron grensen er vist med en prikket linje med pil peker mot exon.
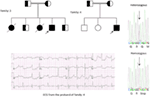
Figur 4. Stamtavle tegning av familien-3 og 4 med autosomal recessiv LQTS2. EKG av proband fra familie-4 er vist under stamtavle tegningen, som viser sinus takykardi, nesten fullstendig av-blokk, bred kompleks rømningsrytme med svært lange qtc-intervaller (QT >600 ms). Mutasjon, c.3208 C > T (S. Q1070X) I kcnh2-genet ble funnet hos pasientene (vist til høyre), merket med en pil.
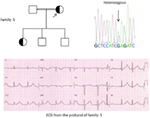
Figur 5. Øverst til venstre: stamtavle av familien-5. Berørt person er vist med fylt sirkel (kvinnelig) Proband er angitt med en pil. 12-bly EKG av proband (i:2). EKG viser en høy topp, bred, stor amplitude T-bølge, qtc-intervall er 580 ms. Screening AV kcnh2-genet viser substitusjon av nukleotid ” G “for en” A ” (c.2362g > a, pil merket), som fører til aminosyresubstitusjon, p. E788K.
Genetiske og kliniske funn i vår studie (12-14) Fra Saudi-Arabia er ganske spennende av flere grunner: (1) totalt har vi undersøkt seks familier, blant dem fire var homozygot / sammensatt heterozygot for mutasjonene og mutasjonene stammer fra en forfedre kilde; (2) alle mutasjonene i LQTS var nye, rapportert bare i Disse Arabiske familier; (3) på grunn av homozygositeten eller sammensatte heterozygositeten for mutasjonene, var kliniske fenotyper også alvorlige i våre studerte familier (12-14). Vi foreslo at de genetiske og fenotypiske observasjonene stammer fra den ekstreme høye graden av consanguineous ekteskap I Saudi-Arabia (108, 109). Vår studie ga det første vitenskapelige beviset om hvilken rolle konsanguinitet har i å utøve en sentral rolle i uforklarlige hjertearytmier og Scd hos barn og ungdom I Saudi-Arabia (12-14). Vi har også vist AT kcnq1-mutasjonen c. 387 -5 T > A (NM_000218) (Figur 3) hadde spredt seg i Assir-provinsen I Saudi-Arabia fra en felles forfedre i flere generasjoner på grunn av høy forekomst av consanguineous ekteskap . Så langt er DETTE den vanligste LQTS1 årsakssammenhengen i Den Saudiarabiske befolkningen, som også ble observert Ved King Faisal Specialist Hospital og Ved Khamis Mashayt Military Hospital (upublisert). Et lignende resultat ble også oppnådd FOR LQTS2 kausal mutasjon, c.3208 C > T (s .Q1070X) (Figur 4) I kcnh2-genet (12, 14), som også er en grunnleggermutasjon I Saudi-Arabia og segregert i mange generasjoner I Assir-regionen (Se kartet, Figur 6). Vi vil forutsi at det finnes et betydelig antall individer med de nevnte forfedre / grunnleggermutasjonene (både I KCNQ1 og KCNH2 OG andre gener) i denne regionen og også i de store byene Som Riyadh, Jeddah og Dammam på grunn av urban migrasjon. Mer grunnlegger eller forfedrenes mutasjoner patogene TIL LQTS er veldig mye tenkt i Andre provinser I Saudi-Arabia på grunn av høy grad av consanguineous ekteskap.
Figur 6. Kart Over Saudi-Arabia(høflighet: Wikipedia). Assir regionen er merket med tykk linje, hvor vi har funnet grunnleggeren mutasjoner I kcnq1 og kcnh2 gener, beskrevet i familie 1-4.
DA LQTS er en autosomal dominant sykdom, forventet vi å observere overveiende heterozygote mutasjonsbærere pasienter. Men i vår studie identifiserte vi hovedsakelig pasienter med recessive mutasjoner (12-14). Klart, disse pasientene blir første symptomatisk og ble hovedsakelig henvist Til Prince Sultan Cardiac Center, bringe dem under vår oppmerksomhet. Likevel innebærer funn fra våre undersøkelser at recessive LQTER kan ha dødelige kliniske fenotyper hos barn, og de er ikke uvanlige I Saudi-Arabia (12-14). Som heterozygote mutasjonsbærere er også utsatt for å utvikle arytmi og dens komplikasjoner, bør samordnet initiativ tas for å bringe de lokale generelle leger, kardiologer og kliniske genetikere i en felles plattform for å identifisere individer i fare. Videre har vi for øyeblikket ingen genetisk informasjon for andre familiære arytmier, FOR EKSEMPEL CPVT, SQTS, BrS, AF, etc. Spesialiserte kardiogenetiske sentre bør ta initiativ til å søke etter de genetiske defekter, mutasjoner, og utføre genotype-fenotype studier i alle former for arvelige arytmier. Det bør også tas i betraktning at ikke alle genetiske arytmier ville ha en familiehistorie, som i mange tilfeller arytmi årsaks mutasjon er de novo opprinnelse, dvs. proband er den første pasienten i den familien med mutasjon og han / hun er kilden til å overføre mutasjon i nedstrøms generasjoner (110, 111). På grunn av den svært høye frekvensen av consanguineous ekteskap I Saudi-Arabia, vi forventer mye mer grunnlegger mutasjoner utøve en avgjørende rolle i medfødte arytmier i dette landet. Å identifisere disse grunnleggermutasjonene bør være vår først og fremst oppgave, noe som vil lette oss i å utvikle en effektiv premarital og også pre-symptomatisk genetisk rådgivning i dette landet. Mutasjoner I LQTS kausale gener kan gi variabilitet i klinisk penetrans, i en ekstrem kan noen bærere antagelig være helt sunne, men noen bærere kan ha sin første manifestasjon av sykdommen som synkope eller plutselig død. Plutselig død av et lite barn eller voksen utgjør en stor psykologisk og følelsesmessig byrde på familien, screening av transportørene er avgjørende da det finnes enkle medisiner, f.eks. Personer med homozygote mutasjoner i LQTS kausale gener kan ha alvorlig morbiditet og ekstrem høy mortalitet inkludert foster brady-takyarytmier, og i mange tilfeller aborter, spontane aborter hos gravide mødre (12-14).
Ikke mye forskning har også blitt utført I Saudi-Arabia om utbredelsen av Ulike SNPs i arytmi knyttet gener og også i gener som regulerer dem. Klaus et al. (112) beskrev en vanlig s1103y-variant I SCN5A-genet assosiert med arytmi hos Afroamerikanere. Varianten allel kalt Y1103 er ansvarlig For å akselerere kanalaktivering, og dermed øke sannsynligheten for hjertearytmier hos Personer Med Afrikansk avstamning (52, 112). K393N er en variant i kcnq1 genet rapportert I LQTS1 pasienter I USA, men I Den Arabiske befolkningen har vi oppdaget denne varianten i 2% av individer(upubliserte data). Hvorvidt K393N-varianten I kcnq1-genet hos Arabere er sammenlignbar MED s1103y (SCN5A) – varianten i Afroamerikanere eller d85n (KCNE1) – varianten i Den Japanske befolkningen, kan også fortjene undersøkelse (59).
Interessekonflikt
forfatterne erklærer at forskningen ble utført i fravær av kommersielle eller økonomiske forhold som kan tolkes som en potensiell interessekonflikt.
Takk
vår oppriktige takknemlighet til Prof. Arthur Wilde, Prof. Connie Bezzina, og utgiveren av tidsskriftet “Heart” for deres tillatelse til å bruke Figur 1 i dette manuskriptet Fra Ref. (113).
1. Bhuiyan ZA. Klinisk Og Genetisk Spektrum Av Arvelige Hjertearytmi Syndromer. Amsterdam: Universitetet I Amsterdam (2009).
2. Priori SG, Aliot E, Blø-Lundqvist C, Bossaert L, Breithardt G, Brugada P, et al. Arbeidsgruppe for plutselig hjertedød, European society of cardiology. Europace (2002) 4:3-18. doi: 10.1053 / eupc.2001.0214
CrossRef Fulltekst
3. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, et al. SCN5A mutasjoner assosiert med en arvelig hjertearytmi, lang QT-syndrom. Celle (1995) 80: 805-11. doi:10.1016/0092-8674(95)90359-3
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
4. Curran MEG, Splawski jeg, Timothy KW, Vincent GM, Grønn ED, Keating MT. Et molekylært grunnlag for hjertearytmi: HERG-mutasjoner forårsaker lang QT-syndrom. Celle (1995) 80: 795-803. doi: 10.1016/0092-8674(95)90358-5
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
5. Wang Q, Curran MEG, Splawski I, Burn TC, Millholland JM, VanRaay TJ, et al. Posisjonell kloning av et nytt kaliumkanalgen: KVLQT1 mutasjoner forårsaker hjertearytmier. Nat Genet (1996) 12:17-23. doi:10.1038 / ng0196-17
Pubmed Abstrakt | Pubmed Fulltekst | CrossRef Fulltekst
6. Lehmann MH, Lehmann MH, Sanguinetti MC, Keating MT. Mutasjoner i hmink-genet forårsaker lang QT-syndrom og undertrykker IKs-funksjonen. Nat Genet (1997) 17:338-40. doi:10.1038 / ng1197-338
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
7. Sanguinetti MC, Jiang C, Curran MEG, Keating MT. En mekanistisk sammenheng mellom en arvet og en ervervet hjertearytmi: HERG koder For IKr kaliumkanalen. Celle (1995) 81: 299-307. doi:10.1016/0092-8674(95)90340-2
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. En ny mutasjon i kaliumkanalgenet KVLQT1 forårsaker Jervell og lange-Nielsen kardioauditory syndrom. Nat Genet (1997) 15:186-9. doi:10.1038 / ng0297-186
Pubmed Abstrakt | Pubmed Fulltekst | CrossRef Fulltekst
9. Kucera JP, Rohr S, Rudy Y. Lokalisering av natriumkanaler i interkalerte disker modulerer hjerteledning. Circ Res (2002) 91:1176-82. doi: 10.1161 / 01.RES.0000046237. 54156. 0 A
Pubmed Abstrakt | Pubmed Fulltekst | CrossRef Fulltekst
10. Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Connexin43 remodeling forårsaket av hemming av plakophilin-2 uttrykk i hjerteceller. Circ Res (2007) 101: 703-11. doi: 10.1161 / CIRCRESAHA.107.154252
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
11. Rohr S. Molekylær crosstalk mellom mekaniske og elektriske veikryss på den interkalerte platen. Circ Res (2007) 101: 637-9. doi: 10.1161 / CIRCRESAHA.107.161901
CrossRef Fulltekst
12. Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, et al. Tilbakevendende intrauterin føtal tap på grunn av nær fravær AV HERG: klinisk og funksjonell karakterisering av en homozygot nonsens HERG Q1070X mutasjon. Hjerte Rytme (2008) 5: 553-61. doi: 10.1016 / j.hrthm.2008.01.020
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
13. Bhuiyan ZA, Momenah TS, Amin TS, Al-Khadra AS, Alder M, Wilde AAM, et al. En Intronisk mutasjon som fører til ufullstendig hopp over ekson-2 i KCNQ1 redder hørsel I jervell og Lange-Nielsen syndrom. Prog Biophys Mol Biol (2008) 98:319-27. doi: 10.1016 / j. pbiomolbio.2008.10.004
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
14. Bhuiyan ZA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi S, Al-Kalaf K, Menns MMAM, et al. Klinisk og genetisk analyse av lang QT-syndrom hos barn fra seks familier I Saudi-Arabia: er de forskjellige? Pediatr Cardiol (2009) 30: 490-501. doi: 10.1007 / s00246-008-9377-y
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
15. Zm, al-Hazzani A, Dzimiri N, Tulbah S, Mallawi Y, Al-Fayyadh M, et al. Identifisering AV en ny kcnq1-mutasjon i En Stor Saudi-familie med langt QT-syndrom: kliniske konsekvenser og forebyggende implikasjoner. Clin Genet (2013) 83:370-4. doi:10.1111/j.1399-0004.2012. 01914.x
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalens av medfødt lang QT-syndrom. Sirkulasjon (2009) 120: 1761-7. doi:10.1161 / SIRKULASJONAHA.109.863209
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
17. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostiske kriterier for lang QT-syndrom. Oppdatering. Sirkulasjon (1993) 88: 782-4. doi: 10.1161 / 01.CIR.88.2.782
CrossRef Fulltekst
18. A. s. g., Wilde AA, H. M., Cho Y., Behr ER, Berul C. et al. SAMMENDRAG: HRS / EHRA / APHRS konsensuserklæring om diagnose og behandling av pasienter med arvelige primære arytmisyndrom. Europace (2013) 15:1389-406. doi: 10.1093 / europace / eut272
CrossRef Full Tekst
19. Liu JF, Jons C, Moss AJ, McNitt S, Peterson DR, Qi M, et al. Syndrom Register. Risikofaktorer for tilbakevendende synkope og påfølgende fatale eller nær fatale hendelser hos barn og ungdom med LANG QT-syndrom. J Am Coll Cardiol (2011) 57: 941-50. doi: 10.1016 / j.jacc.2010.10.025
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
20. J. j. j., J. j., Lehmann MH, Priori S, Robinson J. L., et al. Spektrum av mutasjoner i lang QT-syndrom gener. KVLQT1, HERG, SCN5A, KCNE1 og KCNE2. Sirkulasjon (2000) 102: 1178-85. doi: 10.1161 / 01.CIR.102.10.1178
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
21. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Sammensatte mutasjoner: en vanlig årsak til alvorlig lang QT-syndrom. Sirkulasjon (2004) 109: 1834-41. doi: 10.1161 / 01.CIR.0000125524.34234.13
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
22. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, et al. Risiko for livstruende hjertehendelser blant pasienter med lang QT-syndrom og flere mutasjoner. Hjerte Rytme (2013) 10: 378-82. doi: 10.1016 / j.hrthm.2012.11.006
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
23. J. c., P. S. G., P. P. J., P. R., Ronchetti E. J., Et al. Genetisk testing i lang QT-syndrom: utvikling og validering av en effektiv tilnærming til genotyping i klinisk praksis. JAMA (2005) 294: 2975-80. doi: 10.1001 / jama.294.23.2975
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
24. Roden DM. Klinisk praksis. Lang QT-syndrom. N Engl J Med (2008) 358: 169-76. doi:10.1056 / NEJMcp0706513
Kryssref Full Tekst
25. A. s., A. s., A. S., A. s., A. s., A. s., A. s., A. s., A. s., A. s., A. s., a. s., et al. Genotype-fenotype korrelasjon i lang QT-syndrom: genspesifikke utløsere for livstruende arytmier. Sirkulasjon (2001) 103: 89-95. doi: 10.1161 / 01.CIR.103.1.89
CrossRef Fulltekst
26. Ackerman MJ, Tester DJ, Porter CJ. Svømming, en gen-spesifikk arytmogen utløser for arvet lang QT-syndrom. Mayo Clin Proc (1999) 74: 1088-94. doi:10.4065/74.11.1088
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
27. Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alder M, et al. Genetisk testing for lang QT-syndrom: skille patogene mutasjoner fra godartede varianter. Sirkulasjon (2009) 120: 1752-60. doi:10.1161 / SIRKULASJONAHA.109.863076
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
28. A. J. j., J. J. j., J. j. J., j. J. J., et al. Kliniske aspekter av type-1 lang QT-syndrom etter plassering, kodingstype og biofysisk funksjon av mutasjoner som involverer kcnq1-genet. Sirkulasjon (2007) 115: 2481-9. doi:10.1161 / SIRKULASJONAHA.106.665406
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
29. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. Varianter i 3 ‘ uoversatt region AV Kcnq1-kodet Kv7. 1 kaliumkanal modifiserer alvorlighetsgrad av sykdom hos pasienter med type 1 lang QT-syndrom på en allelspesifikk måte. Eur Hjerte J (2012) 33: 714-23. doi:10.1093 / eurheartj / ehr473
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
30. J. a., J. A., J. A., J. A., J. C., J. W., Et al. Mutasjoner i cytoplasmatiske løkker I kcnq1-kanalen og risikoen for livstruende hendelser: implikasjoner for mutasjonsspesifikk respons på β-blokkerbehandling ved type 1 lang QT-syndrom. Sirkulasjon (2012) 125: 1988-96. doi:10.1161 / SIRKULASJONAHA.111.048041
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
31. J. j. j., J. j. P., J. J. p., J. J. p., J. J. p., J. K., Et al. Jervell og Lange-Nielsen syndrom: naturhistorie, molekylær basis og klinisk utfall. Sirkulasjon (2006) 113:783-90. doi:10.1161 / SIRKULASJONAHA.105.592899
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
32. Bhuiyan ZA, Wilde AA. I hjertet og hørselen kan øret gjøre med mindre enn hjertet. Circ Cardiovasc Genet (2013) 6: 141-3. doi: 10.1161 / CIRCGENETICS.113.000143
Kryssref Fulltekst
33. Wollnik B, Schroeder BC, Kubisch C, Esper HD, Wieacker P, Jentsch TJ. Patofysiologiske mekanismer for dominante OG recessive KVLQT1 K + kanalmutasjoner funnet i arvelige hjertearytmier. Hum Mol Genet (1997) 6:1943-9. doi:10.1093 / hmg / 6.11.1943
Kryssref Fulltekst
34. Wilde AA, Jongbloed RJ, Doevendans PA, Dü DR, Hauer RN, van Langen IM, Et al. Auditiv stimuli som utløser for arytmiske hendelser skiller HERG-relaterte (LQTS2) pasienter fra KVLQT1-relaterte pasienter (LQTS1). J Am Coll Cardiol (1999) 33: 327-32. doi: 10.1016 / S0735-1097(98)00578-6
CrossRef Full Text
35. Aj, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, et al. Økt risiko for arytmiske hendelser i lang QT-syndrom med mutasjoner i poreområdet av den humane ether-a-go-go-relaterte genkaliumkanalen. Sirkulasjon (2002) 105: 794-9. doi:10.1161 | hc0702.105124
Pubmed Abstrakt | Pubmed Fulltekst / CrossRef Fulltekst
36. Lupoglazoff JM, Denjoy I, Skurk E, Fressart V, Simon F, Bozio A, et al. Lang QT-syndrom hos nyfødte: ledningsforstyrrelser assosiert MED herg-mutasjoner og sinusbradykardi med kcnq1-mutasjoner. J Am Coll Cardiol (2004) 43: 826-30. doi: 10.1016 / j.jacc.2003.09.049
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
37. Chevalier P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ, et al. Torsades de pointes kompliserer atrioventrikulær blokk: bevis for en genetisk predisposisjon. Hjerte Rytme (2007) 4: 170-4. doi: 10.1016 / j.hrthm.2006.10.004
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
38. Hoorntje T, Alder M, van Tintelen P, van Der Lip K, Sreeram N, van Der Wal A, et al. Homozygot for tidlig avkorting AV herg-proteinet: human HERG knockout. Sirkulasjon (1999) 100: 1264-7. doi: 10.1161 / 01.CIR.100.12.1264
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
39. Piippo K, Laitinen P, Svane H, Toivonen L, Viitasalo M, Pasternack M, et al. Homozygositet for en herg kalium kanal mutasjon forårsaker en alvorlig form for lang QT-syndrom: identifisering av en tilsynelatende grunnlegger mutasjon I Finnene. J Am Coll Cardiol (2000) 35: 1919-25. doi: 10.1016 / S0735-1097(00)00636-7
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
40. Johnson Wh Jr, Yang P, Yang T, Lau YR, Mostella BA, Wolff DJ, et al. Klinisk, genetisk og biofysisk karakterisering av en homozygot HERG-mutasjon som forårsaker alvorlig neonatal lang QT-syndrom. Pediatr Res (2003) 53: 744-8. doi: 10.1203 / 01.PDR.0000059750.17002.B6
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
41. Ackerman MJ, Tester DJ, Jones GS, Vil ML, Burrow CR, Curran MEG. Etniske forskjeller i hjertekaliumkanalvarianter: implikasjoner for genetisk følsomhet for plutselig hjertedød og genetisk testing for medfødt lang QT-syndrom. Mayo Clin Proc (2003) 78: 1479-87. doi:10.4065/78.12.1479
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
42. C, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T er en genetisk modifier av latent medfødt lang QT-syndrom. Sirkulasjon (2005) 112: 1251-8. doi:10.1161 / SIRKULASJONAHA.105.549071
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
43. Nof E, Cordeiro Jm, P Hryvrez GJ, Scornik FS, Calloe K, Kjærlighet B, et al. En vanlig enkelt nukleotidpolymorfisme kan forverre lang QT type 2 syndrom som fører til plutselig barnedød. Circ Cardiovasc Genet (2010) 3: 199-206. doi: 10.1161 / CIRCGENETICS.109.898569
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
44. Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, Et al. En Enkelt Na ( + ) kanal mutasjon forårsaker både lang QT og brugada syndromer. Circ Res (1999) 85:1206-13. doi: 10.1161 / 01.RES.85.12.1206
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
45. C, C, C, C, C, C, C, C, Et al. Novel SCN5A mutasjon fører enten til isolert hjerteledning defekt eller Brugada syndrom i en stor fransk familie. Sirkulasjon (2001) 104: 3081-6. doi:10.1161 / hc5001.100834
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
46. M. m., m. m., M. m., M. m., M. M., M. M., M. m., m. m., m. m., m. m., m. m., et al. E1784k-mutasjonen I SCN5A er assosiert med blandet klinisk fenotype av type 3 lang QT-syndrom. J Clin Invest (2008) 118:2219-29. doi:10.1172 / JCI34057
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
47. Keller DI, Acharfi S, Delacr ④az E, Benammar N, Rotter M, Pfammatter JP, et al. En ny mutasjon I SCN5A, delQKP 1507-1509, forårsaker lang QT-syndrom: Rolle q1507 rester i natriumkanalinaktivering. J Mol Celle Cardiol (2003) 35:1513-21. doi:10.1016 / j. yjmcc.2003.08.007
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
48. Sg, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risikostratifisering i lang QT-syndromet. N Engl J Med (2003) 348: 1866-74. doi:10.1056 / NEJMoa022147
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
49. J. M. J. M., Chav T., Baroudi G., Berthet M., Denjoy i., Cauchemez B., et al. Homozygot SCN5A-mutasjon i lang QT-syndrom med funksjonell to-til-en atrioventrikulær blokk. Circ Res (2001) 89: E16-21. doi: 10.1161 / hh1401.095087
Kryssref Full Tekst
50. Shi R, Zhang Y, Yang C, Huang C, Zhou X, Qiang H, et al. Hjertenatriumkanalmutasjonen delQKP 1507-1509 er assosiert med det ekspanderende fenotypiske spekteret AV LQT3, ledningsforstyrrelse, dilatert kardiomyopati og høy forekomst av ungdoms plutselig død. Europace (2008) 10:1329-35. doi: 10.1093 / europace / eun202
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
51. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartscheer A, Eckardt L, Paul M, et al. En heterozygot delesjonsmutasjon I natriumkanalgenet scn5a MED tap – og gevinst-av-funksjonsegenskaper manifesterer seg som isolert ledningssykdom, uten tegn På Brugada eller lang QT-syndrom. PLoS One (2013) 8: e67963. doi:10.1371 / tidsskrift.pone.0067963
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
52. Plant LD, Bowers PN, Liu Q, Morgan T, Zhang T, Stat MW, Et al. En vanlig kardial natriumkanalvariant assosiert med plutselig barnedød hos Afroamerikanere, SCN5A S1103Y. J Clin Invest (2006) 116:430-5. doi: 10.1172 / JCI25618
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
53. Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, et al. Ankyrin-b-mutasjon forårsaker type 4 lang QT hjertearytmi og plutselig hjertedød. Natur (2003) 421: 634-9. doi: 10.1038 / nature01335
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
54. Schott JJ, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB, et al. Kartlegging av et gen for lang QT-syndrom til kromosom 4q25-27. Am J Hum Genet (1995) 57:1114-22.
Pubmed Abstrakt | Pubmed Fulltekst
55. Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 og lsK (minK) proteiner forbinder for å danne i (Ks) hjertekaliumstrømmen. Natur (1996) 384: 78-80. doi:10.1038/384078a0
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
56. A. j., A. J., A. J., A. J., A. J., A. j., A. j., A. j., a. j., a. j., a. j., et al. Koassembly AV K (V)LQT1 og minK (IsK) proteiner for å danne hjerte i (Ks) kaliumkanal. Natur (1996) 384: 80-3. doi: 10.1038 / 384080a0
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
57. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al. KCNE1 mutasjoner forårsaker Jervell og Lange-Nielsen syndrom. Nat Genet (1997) 17: 267-8. doi:10.1038 / ng1197-267
Kryssref Full Tekst
58. Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutasjon av genet for IKs assosiert med Både Jervell og Lange-Nielsen og Romano-Ward former for lang QT-syndrom. Sirkulasjon (1998) 97: 142-6. doi: 10.1186/1471-2350-9-24
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85N, EN kcne1 polymorfisme, er en sykdomsfremkallende genvariant i lang QT-syndrom. J Am Coll Cardiol (2009) 54: 812-9. doi: 10.1016 / j.jacc.2009.06.005
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
60. Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, et al. Genetiske variasjoner AV KCNQ1, KCNH2, SCN5A, KCNE1 og KCNE2 hos legemiddelinduserte pasienter med LANG QT-syndrom. Mol Med (2004) 82:182-8. doi: 10.1007 / s00109-003-0522-z
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
61. Abbott GW, Sesti F, Splawski I, Buck MEG, Lehmann MH, Timothy KW, et al. MiRP1 danner IKr kaliumkanaler med HERG og er forbundet med hjertearytmi. Celle (1999) 97: 175-87. doi:10.1016 / S0092-8674 (00)80728-X
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
62. Gordon E, Panaghie G, Deng L, Bee KJ, Roepke TK, Krogh-Madsen T, Et al. En KCNE2-mutasjon hos en pasient med hjertearytmi indusert av auditiv stimuli og serumelektrolyttbalanse. Cardiovasc Res (2008) 77: 98-106. doi:10.1093/cvr | cvm030
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
63. Plaster NM, Tawil R, Tristani-Firouzi M, Kanú S, Bendahhou S, Tsunoda A, et al. Mutasjoner I Kir2.1 forårsaker utviklingsmessige og episodiske elektriske fenotyper Av Andersens syndrom. Celle (2001) 105: 511-9. doi: 10.1016 / S0092-8674(01)00342-7
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primær struktur og funksjonell uttrykk for en mus innover likeretter kaliumkanal. Natur (1993) 362: 127-33. doi:10.1038/362127a0
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
65. Raab-Graham KF, Radeke CM, Vandenberg CA. Molekylær kloning og uttrykk for et menneskelig hjerte innover likeretter kaliumkanal. Neuroreport (1994) 5: 2501-5. doi: 10.1097/00001756-199412000-00024
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
66. Jørgensdatter I, Jørgensdatter LM, Jørgensdatter lm, Jørgensdatter lm, Jørgensdatter lm, Jørgensdatter lm, et al. Ca (V) 1.2 kalsiumkanal dysfunksjon forårsaker en multisystem lidelse inkludert arytmi og autisme. Celle (2004) 119: 19-31. doi: 10.1016 / j.celle.2004.09.011
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
67. Jørgensdatter I, Jørgensdatter I, Jørgensdatter i, Jørgensdatter i, Jørgensdatter i, Jørgensdatter i, Jørgensdatter i, Et al. Alvorlig arytmieforstyrrelse forårsaket av kardiale l-type kalsiumkanalmutasjoner. Proc Natl Acad Sci Usa (2005) 102: 8089-96. doi: 10.1073 / pnas.0502506102
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
68. Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Brutto G, Turner L, et al. Long QT, syndactyly, felles kontrakturer, slag og roman cacna1c mutasjon: utvide spekteret Av Timothy syndrom. Er J Med Genet A (2012) 158A:182-7. doi: 10.1002 / ajmg.a.34355
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
69. Duphendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. Mors mosaicism forvirrer neonatal diagnose av Type 1 Timothy syndrom. Pediatri (2013) 131: e1991-5. doi: 10.1542 / peds.2012-2941
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
70. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. Mutant caveolin-3 induserer vedvarende sen natriumstrøm og er assosiert med lang QT-syndrom. Opplag (2006) 114:2104–12. doi:10.1161 / SIRKULASJONAHA.106.635268
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
71. Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, Et al. Ny mekanisme for plutselig barnedødssyndrom: vedvarende sen natriumstrøm sekundær til mutasjoner i caveolin-3. Hjerte Rytme (2007) 4: 161-6. doi: 10.1016 / j.hrthm.2006.11.030
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
72. Engelman JA, Zhang X, Galbiati F, Volonte D, Sotgia F, Pestell RG, et al. Molekylær genetikk av caveolin-genfamilien: implikasjoner for menneskelig kreft, diabetes, Alzheimers sykdom og muskeldystrofi. Am J Hum Genet (1998) 63:1578-87. doi:10.1086/302172
CrossRef Full Text
73. Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Lokalisering av hjerte L-Type Ca(2+) kanaler til et kaveolært makromolekylært signalkompleks er nødvendig for beta (2) – adrenerg regulering. Proc Natl Acad Sci Usa (2006) 103: 7500-5. doi: 10.1073 / pnas.0503465103
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
74. Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, et al. SCN4B-kodet natriumkanal beta4-underenhet ved medfødt lang QT-syndrom. Sirkulasjon (2007) 116: 134-42. doi:10.1161 / SIRKULASJONAHA.106.659086
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
75. Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutasjon Av et a-kinase-forankringsprotein forårsaker lang QT-syndrom. Proc Natl Acad Sci Usa (2007) 104:20990-5. doi: 10.1073 / pnas.0710527105
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
76. Wu G, Ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, et al. Alfa-1-syntrofin mutasjon og lang QT-Syndrom: en sykdom av natrium kanal avbrudd. Circ Arrhythm Elektrofysiol (2008) 1: 193-201. doi: 10.1161 / CIRCEP.108.769224
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnen M, et al. Identifikasjon Av En Kir3.4 mutasjon i medfødt lang QT-syndrom. Er J Hum Genet (2010) 86: 872-80. doi: 10.1016 / j.ajhg.2010.04.017
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
78. Roden DM. Mekanismer og behandling av proarytmi. Am J Cardiol (1998) 82:49i–57i. doi:10.1016 / S0002-9149(98)00472-X
Kryssref Full Tekst
79. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. Et strukturelt grunnlag for legemiddelinducert lang QT-syndrom. Proc Natl Acad Sci Usa (2000) 97: 12329-33. doi: 10.1073 / pnas.210244497
Kryssref Full Tekst
80. Kannankeril PJ, Roden DM. Drug-indusert lang QT og torsade de pointes: nylige fremskritt. Curr Opin Cardiol (2007) 22:39-43. doi: 10.1097 / HCO.0b013e32801129eb
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
81. Veerman CC, Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC, et al. Langsom forsinket likeretterkaliumstrømblokkade bidrar viktigere til legemiddelinducert lang QT-syndrom. Circ Arrhythm Elektrofysiol (2013) 6: 1002-9. doi: 10.1161 / CIRCEP.113.000239
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
82. Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. En vanlig polymorfisme assosiert med antibiotisk-indusert hjertearytmi. Proc Natl Acad Sci Usa (2000) 97: 10613-8. doi: 10.1073 / pnas.180223197
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
83. Kää s, Crawford Dc, synder Mf, behr Er, Kannankeril Pj, wilde Aa, et al. En stor kandidatgenundersøkelse identifiserer kcne1 D85N polymorfisme som en mulig modulator av legemiddelinducerte torsades de pointes. Circ Cardiovasc Genet (2012) 5: 91-9. doi: 10.1161 / CIRCGENETICS.111.960930
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
84. K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase S, Et al. Anti-KCNH2 antistoff-indusert lang QT-syndrom: ny ervervet form av lang QT-syndrom. J Am Coll Cardiol (2007) 50: 1808-9. doi: 10.1016 / j.jacc.2007.07.037
CrossRef Fulltekst
85. Aj, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, et al. EKG T-bølge mønstre i genetisk forskjellige former for arvelig lang QT-syndrom. Sirkulasjon (1995) 92: 2929-34. doi: 10.1161 / 01.CIR.92.10.2929
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
86. Jørgensen L, Jørgensen L, Jørgensen L, Jørgensen l, et al. Spektrum AV ST-t-bølgemønstre og repolariseringsparametere ved medfødt lang QT-syndrom: EKG-funn identifiserer genotyper. Sirkulasjon (2000) 102: 2849-55. doi: 10.1161 / 01.CIR.102.23.2849
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
87. T. H. H. L., Bardai A, Shimizu W, Moss AJ, Schulze-Bahr E, Noda T, Et al. Genotype-spesifikk utbrudd av arytmier i medfødt lang QT-syndrom: mulige terapiimplikasjoner. Sirkulasjon (2006) 114: 2096-103. doi:10.1161 / SIRKULASJONAHA.106.642694
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
88. B EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Alder-og kjønnsrelaterte forskjeller i kliniske manifestasjoner hos pasienter med medfødt lang QT-syndrom: funn Fra Det Internasjonale Lqts-Registeret. Sirkulasjon (1998) 97: 2237-44. doi: 10.1161 / 01.CIR.97.22.2237
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
89. Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, et al. Påvirkning av genotype på det kliniske løpet av lang QT-syndromet. International Long-QT Syndrome Registry Research Group (Engelsk). N Engl J Med (1998) 339: 960-5. doi: 10.1056 / NEJM199810013391404
Pubmed Abstrakt / Pubmed Full Tekst / CrossRef Full Tekst
90. J. j. j., j. j., J. j., J. J., J. J., J. J., J. J., J. J., J. J., J. J., Et al. Lang QT-syndrom etter 40 år. Sirkulasjon (2008) 117: 2192-201. doi:10.1161 / SIRKULASJONAHA.107.729368
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
91. Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, et al. Syndrom Register. Modulerende effekter av alder og kjønn på det kliniske forløpet av lang QT-syndrom etter genotype. J Am Coll Cardiol (2003) 42: 103-9. doi: 10.1016 / S0735-1097(03)00554-0
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
92. Seth R, Moss AJ, McNitt S, Zareba W, Andrews ML, Qi M, Et al. Lang QT-syndrom og graviditet. J Am Coll Cardiol (2007) 49: 1092-8. doi: 10.1016 / j.jacc.2006.09.054
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
93. Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, et al. En molekylær sammenheng mellom sudden infant death syndrome og long-QT syndrom. N Engl J Med (2000) 343: 262-7. doi:10.1056 / NEJM200007273430405
Kryssref Full Tekst
94. Pj, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, et al. Molekylær diagnose hos et barn med plutselig barnedødssyndrom. Lancet (2001) 358:1342-3. doi: 10.1016 / S0140-6736(01)06450-9
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
95. Christiansen M, Tø N, Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutasjoner I HERG K + – ion-kanalen: en ny sammenheng mellom lang QT-syndrom og plutselig barnedødssyndrom. Am J Cardiol (2005) 95: 433-4. doi:10.1016 / j. amjcard.2004.09.054
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
96. Tester DJ, Ackerman MJ. Postmortem lang QT-syndrom genetisk testing for plutselig uforklarlig død hos unge. J Am Coll Cardiol (2007) 49: 240-6. doi: 10.1016 / j.jacc.2006.10.010
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
97. Hofman N, Tan HL, Clur SA, Alder M, van Langen IM, Wilde AA. Bidrag av arvelig hjertesykdom til plutselig hjertedød i barndommen. Pediatri (2007) 120: e967-73. doi: 10.1542 / peds.2006-3751
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
98. J. j. j. p., Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T., et al. De novo mutasjon I scn5a genet assosiert med tidlig debut av krybbedød. Sirkulasjon (2001) 104: 1158-64. doi: 10.1161 / hc3501.095361
Kryssref Full Tekst
99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, et al. Kardial natriumkanal dysfunksjon i plutselig barnedødssyndrom. Sirkulasjon (2007) 115: 368-76. doi:10.1161 / SIRKULASJONAHA.106.646513
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
100. Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, Et al. Molekylær og funksjonell karakterisering av nye glyserol-3-fosfat dehydrogenase 1 som gen (GPD1-L) mutasjoner i krybbedød. Sirkulasjon (2007) 116: 2253-9. doi: 10.1161 / SIRKULASJONAHA.107.704627
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
101. Tan BH, Pundi KN, Van Norstrand DW, Valdivia Cr, Tester DJ, Medeiros-Domingo A, et al. Sudden infant death syndrome-assosierte mutasjoner i natriumkanal beta subenheter. Hjerte Rytme (2010) 7: 771-8. doi: 10.1016 / j.hrthm.2010.01.032
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
102. Miller TE, Estrella E, Myerburg RJ, Garcia De Viera J, Moreno N, Rusconi P, et al. Tilbakevendende tredje trimester føtalt tap og mors mosaikk for lang QT-syndrom. Sirkulasjon (2004) 109: 3029-34. doi: 10.1161 / 01.CIR.0000130666.81539.9 E
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
103. Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van Der Heijden JF, et al. Ikke alle betablokkere er like i forvaltningen av lang QT-syndrom type 1 og 2: høyere tilbakefall av hendelser Under Metoprolol. J Am Coll Cardiol (2012) 60: 2092-6. doi: 10.1016 / j.jacc.2012.07.046
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
104. A. p. J., P. J. S., C. J., C. J., C. J., C. j., c. j., C. j., c. j., c. j., C. j., C. j., Et al. Venstre hjerte sympatisk denervering i styringen av høyrisikopasienter påvirket av lang QT-syndromet. Sirkulasjon (2004) 109: 1826-33. doi: 10.1161 / 01.CIR.0000125523.14403.1 E
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
105. Singh B, al Shahwan SA, Habbab MA, al Deeb SM, Biary N. Idiopatisk lang QT-syndrom: spør det rette spørsmålet. Lancet (1993) 341:741. doi:10.1016/0140-6736(93)90501-7
CrossRef Full Text
106. Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. det lange QT-syndromet med nedsatt atrioventrikulær ledning: en ondartet variant hos spedbarn. J Cardiovasc Elektrofysiol (1998) 9:1225-32. doi:10.1111/j.1540-8167.1998.tb00096.x
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
107. Kantoch MJ, Qurashi MM, Bulbul ZR, Gorgels AP. En nyfødt med en kompleks medfødt hjertesykdom, atrioventrikulær blokk, og torsade de pointes ventrikkeltakykardi. Pacing Clin Elektrofysiol (1998) 21: 2664-7. doi: 10.1111 / j.1540-8159. 1998.tb00043.x
Kryssref Fulltekst
108. Al-Hazmi MA, Al-Swailem AR, Krigsherjede AS, Al-Swailem AM, Sulaimani R, Al-Meshari AA. Slektskap blant Den Saudiarabiske befolkningen. J Med Genet (1995) 32: 623-6. doi:10.1136 / jmg.32.8.623
Kryssref Fulltekst
109. El Mouzan MI, AL Salloum AA, Al Herbish AS, Qurachi MM, Al OMAR AA. Konsanguinitet og store genetiske lidelser hos Saudiske barn: en samfunnsbasert tverrsnittsstudie. Ann Saudi Med (2008) 28:169-73. doi:10.4103/0256-4947.51726
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
110. Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van Tintelen JP, et al. Ryr2-kodet ryanodinreseptor / kalsiumfrigjøringskanal hos pasienter diagnostisert med enten katekolaminerg polymorf ventrikulær takykardi eller genotype negativ, treningsindusert LANG QT-syndrom: en omfattende åpen leseramme mutasjonsanalyse. J Am Coll Cardiol (2009) 54: 2065-74. doi: 10.1016 / j.jacc.2009.08.022
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
111. Al – Aama JY, Al-Ghamdi S, Bdier AY, Wilde AA, Bhuiyan ZA. De novo-mutasjon I kcnq1-genet som er årsak til Jervell Og Lange-Nielsens Syndrom. Clin Genet (2013). doi: 10.1111 / cge.12300
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst
112. I. l., T. I., T. I., T. I., T. I., T. I., T. I., t. I. M., T. I. M., T. I. A., Et al. Variant AV SCN5A natriumkanal involvert i risiko for hjertearytmi. Vitenskap (2002) 297: 1333-6. doi:10.1126 / vitenskap.1073569
Kryssref Fulltekst
113. Wilde AA, Bezzina CR. Genetikk av hjertearytmier. Hjerte (2005) 91: 1352-8.