Mukopolysakkaridoser: tidlige diagnostiske tegn hos spedbarn og barn
Mukopolysakkaridoser (Mps) er en gruppe klinisk heterogene sykdommer forårsaket av mangler i lysosomale enzymer som kreves for nedbrytning av glykosaminoglykaner (GAGs).
MPS er preget av progressive og systemiske kliniske manifestasjoner. Til tross for deres biokjemiske og genetiske heterogenitet, deler forskjellige typer viktige kliniske trekk i varierende kombinasjoner, inkludert ledd-og skjelettdysplasi med stivhet (unntatt MPS IV hvor det er slapphet) og smerte, grove ansiktsegenskaper, hornhinneklyng, inguinal eller abdominal brokk, tilbakevendende øvre luftveisinfeksjoner, hjerteklaffsykdom, karpaltunnelsyndrom og variabel nevrologisk involvering. Disse funksjonene vises vanligvis i de første månedene av livet for de alvorlige former og i tidlig barndom for de mer svekkede former, men blir ofte undervurdert og generelt tatt i betraktning bare når det er tydelig.
sjeldenheten av disse lidelsene og variabiliteten i klinisk presentasjon fører ofte til en diagnostisk forsinkelse; dette kan variere fra måneder, for de alvorlige formene, til år, for de svekkede (se Rigoldi et al. i dette tillegget ). Likevel, på grunn av den raske utviklingen og det presserende behovet for intervensjon i de alvorlige presentasjonene, kan selv måneder med forsinkelse i diagnosen gi katastrofale resultater for pasientens fremtidige helse.
vårt mål er å understreke i denne gjennomgangen de svært tidlige tegnene på de alvorlige formene som bør varsle barnelege om deres første utseende.
hvilke tegn / symptomer kan vi forvente hos barn med alvorlige FORMER for MPS?
Symptomer og tegn som tyder på ulike former FOR MPS er rapportert I Tabell 1 etter alder. I løpet av de første 6 månedene av livet kan man se inguinal brokk, unormalt hyppige luftveisinfeksjoner, otitis og organomegali hos MPS-pasienter, spesielt I Mps I (Hurler syndrom) som har det mest alvorlige utbruddet. Videre, fra 6 til 12 måneder, utvikler disse spedbarnene ofte en gibbus (toraco-lumbale kyphos), hjerteklump, navlestreng, mild hypotoni og vekstforsinkelse . De kan også ha det typiske grove ansiktsutseendet (Fig. 1). MPS II, VI og VII kan ha en lignende tidlig fenotype. MPS IV-pasienter har en tidlig skjelettfenotype med utseende av hoftedysplasi i de første månedene av livet og duebrystet (pectus carinatum) i de første 12-18 månedene. MPS ii spedbarn tilstede med en lignende involvering, men med senere debut. I det andre året av livet, mps I Hurler syndrom spedbarn utvikle kognitiv forsinkelse mens EN MPS II pasient kan identifiseres bare på grunn av overvekst, hyppige luftveisinfeksjoner, og flere operasjoner ; de typiske ansiktstrekk vises bare senere. NOEN MPS II-pasienter utvikler også hyperaktivitet mellom 18 og 24 måneder. I det tredje år av livet er hyperaktivitet og kognitiv forsinkelse lett gjenkjennelig i MPS II OG MPS III.
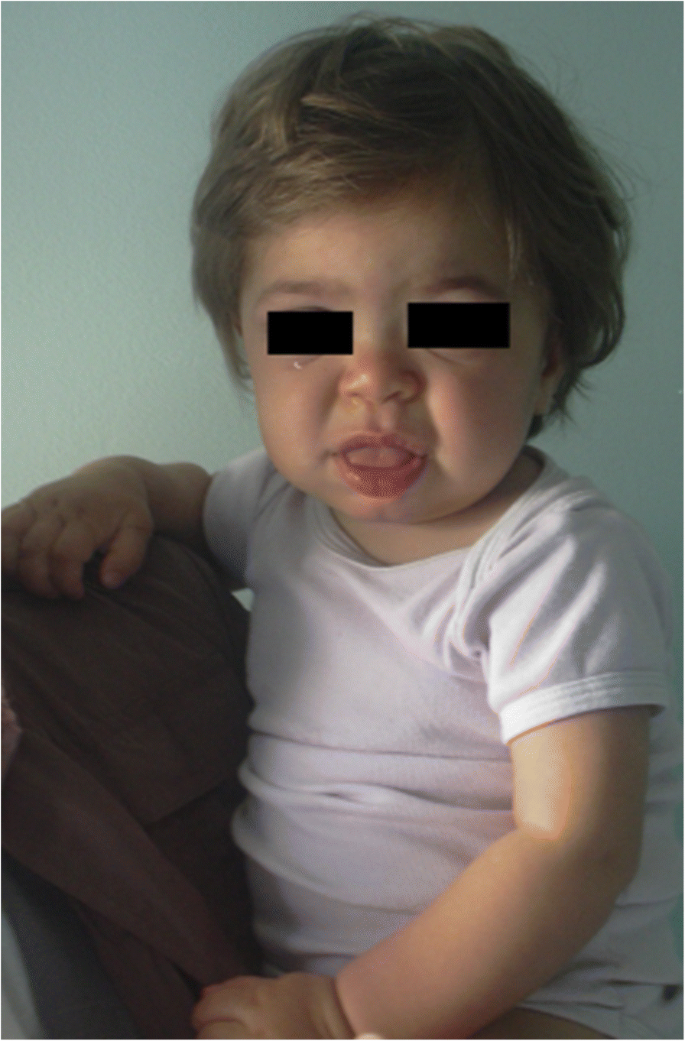
MPS IH i 1 ar gammel pasient. Grove facies: flat nesebro, makroglossi, frontal bossing
oppsummert er skjelett manifestasjoner typiske og noen ganger forgjengelige. Grov facies og kognitiv forsinkelse er ikke tidlige fremtredende tegn.
hva er de forskjellige presentasjonene av DE ulike TYPENE PARLAMENTSMEDLEMMER?
MPS med somatisk og kognitiv involvering
Mukopolysakkaridose type i (MPS i; OMIM #252800) (Fig. 1, 2, 3, 4 og 5) skyldes en mangel på den lysosomale hydrolasen α-l-iduronidase, noe som fører til akkumulering av to GAGs i flere systemer: dermatansulfat (DS) og heparansulfat (hs) . BASERT på alvorlighetsgraden er MPS i tradisjonelt delt inn i tre kliniske fenotyper; disse formene representerer imidlertid et kontinuum av sykdoms alvorlighetsgrad. Hurler syndrom, den mest alvorlige formen, presenterer vanligvis i løpet av det første år av livet. Berørte barn utvikler raskt betydelig kognitiv svekkelse og somatisk sykdom i flere organsystemer, noe som fører til døden innen det første tiåret uten behandling. De svekkede former AV MPS i, kjent som Hurler–Scheie syndrom og Scheie syndrom, henholdsvis, er preget av senere utbruddet av symptomer, lengre levealder, og mild eller ingen sentralnervesystemet (CNS) involvering . Arv er autosomal recessiv, og i minst 50% av tilfellene er fenotypeforutsigelse mulig på grunnlag av genotype, mens i de resterende 50% oppdages nye mutasjoner . Prevalens er ca. 1 av 100 000 levendefødte .

MPS IH. En Gibbus i en 9 måneder gammel pasient. b Ryggrad røntgen av ryggraden i en 3 år gammel pasient. c T2-vektet magnetisk resonansbilde av ryggraden som viser signifikant kypose og vertebralkanalstenose

MPS IH. Røntgen bevis på dysostose multiplex. En Fortykkelse av ribbeina i en 3 år gammel pasient og b hofteleddsdysplasi hos en annen pasient i alderen 18 måneder

MPS IH: En Klo hånd i en 18 måneder gammel pasient; b Hånd X-ray.

MPS IH. En Fortykkelse av den kortikale bein i skallen og unormal “J-formet” konformasjon av sella turcica. Dilatasjon av de periventrikulære rom i B TEFT og c T-2 vektet magnetisk resonans bilder av en pasient i alderen 17 måneder
Mukopolysakkaridose TYPE II (MPS II; omim #309900) (Fig. 6), også kjent Som Hunters syndrom, er et resultat av mangel på enzymet iduronat-2-sulfatase (I2S) med påfølgende GAG akkumulering AV DS og HS . Prevalens er rundt 1 av 140.000 – 156.000 mannlige levendefødte . Spesielt er dette den eneste X-koblede MPS, og kvinnelige bærere er vanligvis asymptomatiske; imidlertid kan de bli eksepsjonelt påvirket på grunn Av abnormiteter I X-kromosomet, homozygositet eller skjev-x-inaktivering . Det fenotypiske uttrykket spenner også over et bredt spekter av klinisk alvorlighetsgrad. I alvorlige tilfeller eksisterer markert progressiv nevrologisk involvering og somatisk sykdom, og døden oppstår vanligvis i det andre tiåret av livet. Andre pasienter med minimal eller ingen nevrologisk dysfunksjon kan presentere en alvorlig somatisk involvering, mens i motsatt ende av spekteret er det pasienter med bare minimal somatiske manifestasjoner og normal intelligens som overlever i voksen alder . Tidlige tegn og symptomer deles av de alvorlige formene FOR MPS I OG II.

MPS II. En 3 år gammel pasient med bare milde karakteristiske ansiktsegenskaper
Sporadiske tilfeller av hydrops fetalis er beskrevet I MPS I, selv om, generelt, disse barna vises normalt ved fødselen. De første tegnene på MPS i vises vanligvis i de aller første månedene av livet, mens DE I MPS II vanligvis oppstår litt senere . Kiely et al. i en retrospektiv studie med 55 Hurler-pasienter, ble det observert at respiratoriske symptomer, spisevansker, inguinal brokk og otitis media hadde oppstått ved en median alder ≤ 6 måneder, og kyphos, hjerteavvik, forstørret hodeomkrets, hypotoni, organomegali, hornhinneklyng, leddbegrensning og navlestreng oppstod mellom 6 og 12 måneder. Umbilical brokk er en av de tidligste presentere funksjonene I MPS i OG II, og inguinal brokk er rapportert hos ca 60% av pasientene .
En annen vanlig første ledetråd for MPS I OG II kan være øvre luftveisinfeksjoner med økte sekreter. Hyppig otitis, kronisk tilbakevendende rhinitt og vedvarende neseutslipp uten åpenbar infeksjon er ikke spesifikke, men kan bidra til å styrke den diagnostiske mistanken hvis den er forbundet med mer spesifikke tegn. Lagring AV GAG i orofarynx fører til utvidelse av mandler og adenoider, og bidrar til øvre luftveiskomplikasjoner. Et annet hyppig funn er støyende pust, spesielt om natten, assosiert i senere stadier med obstruktiv søvnapne. Som følge av hyppige mellomøreinfeksjoner og dysostose i mellomøret i mellomøret, er hørselstap hyppig, med både ledende og sensorineurale underskudd . Tracheobronchomalacia er ofte observert og kan føre til akutt luftveisobstruksjon eller kollaps, med dette å være en av de viktigste årsakene til død i senere år .
Adenoidektomi, mandlene, og tympanostomi, sammen med brokk reparasjon, er hyppigere og tidlig kirurgiske inngrep FOR mps i og II pasienter, ofte utført før vite diagnosen (i mer enn 50% av tilfellene).
Tilbakevendende diare er ganske vanlig i tidlige STADIER av MPS I, og kan også være tilstede hos mps II-pasienter med nevrologisk involvering, mens det er uvanlig hos de mildt berørte pasientene .
Gibbus misdannelse (dorso-lumbale kyfose) (Fig. 2) blir klinisk tydelig ved en median alder på 8.6 måneder til 1 år I MPS I, som representerer en ganske spesifikk og forgjengelig markør FOR MPS generelt. Vertebrale legemer ser ut til å være flate og nebbete, noe som potensielt kan føre til senere komplikasjoner som spinal nerve entrapment, akutt spinal skade og atlanto-occipital ustabilitet; derfor må det utvises spesiell forsiktighet for å forhindre dislokasjon av atlanto-aksial ledd under generell anestesi og kirurgi. Etter 1 års alder representerer leddkontrakturer og genu valgum andre hyppige tegn på leddhemming og progressiv skjelettdysplasi, som lett kan påvises tidligere enn 2 år hos alle spedbarn som er rammet av MPS. Alle disse skjelett manifestasjoner sammen, typisk FOR MPS, er oppkalt dysostosis multiplex. Fortykning av ribbenene kan ses på røntgenbilder (Fig. 3a), de lange beinene er korte med brede aksler, og knærne er utsatt for valgus og varus deformiteter. Phalangeal dysostosis og synovial jevning føre til en karakteristisk klo hånd misdannelse (Fig. 4), som er en annen typisk funksjon som kan øke mistanke om sykdommen. Hofteleddsdysplasi er ofte grunnen til at disse barna kan først bli sett av ortopediske spesialister. Vanligvis er bekkenet dårlig dannet, lårhodene er små, og coxa valga er vanlig, med progressiv og svekkende hoftedeformitet (Fig. 3b) .
det er en merkbar forskjell MELLOM MPS I OG MPS II; i mps i avtar skjelettveksten med 3 år , mens mps II-pasienter viser overvekst i de første 5-6 årene, og senker veksten deretter .
Grovdannelse av ansiktsfunksjonene (bred nese med flared nesebor, fremtredende supraorbital rygger, store avrundede kinn, tykke lepper, forstørret utstående tunge) forårsaket av lagring Av GAGs i det myke vevet i orofacialområdet og ansiktsbenene blir tydelig innen de første 2 årene, i en median alder på 1,1 år for Hurler sykdom og mellom 18 måneder og 4 år i den alvorlige Formen For Hunter sykdom , selv om subtile fenotypiske endringer kan oppdages tidligere (så tidlig som i andre halvdel av det første året) av pediatrikere med spesifikk erfaring (fig. 1). Makrocefali blir gradvis mer tydelig, og scaphocefaly er vanlig (Fig. 5), men mikrocefali er også mulig hos Hurler-pasienter . Ansikts-og kroppshirsutisme er alltid tydelig i en alder av 24 måneder . Huden kan være tykk og uelastisk. Spesielt utvikler NOEN MPS II-pasienter en særegen hudlesjon, som er beskrevet som elfenbenhvite papiller (pebble-like) på øvre rygg og sider av overarmene, patognomonisk Av Hunters syndrom .
Kardiomyopati og progressiv fortykkelse og stivning av ventilbladene hos MPS I-pasienter kan føre til mitral og, sjeldnere, aortaregurgitasjon . En murmur kan oppdages i de første månedene av livet og har noen ganger blitt rapportert som det første tegn på sykdommen . Hjerteinnblanding er også tilstede hos nesten alle pasienter MED MPS II, men dette starter ved rundt 5 års alder . Ventil sykdom kan føre til høyre og venstre ventrikkel hypertrofi og hjertesvikt.
Fremspring i magen forårsaket av progressiv hepatosplenomegali er lett tydelig etter 6 måneders alder, selv om lagring Av GAGs i leveren og milten ikke fører til organdysfunksjon .
hornhinneklynging forekommer hos nesten alle personer med MPS I ved tidligere enn 15 måneders alder, noe som potensielt forårsaker alvorlig synshemming. Tvert imot er hornhinnen ikke et typisk trekk Ved Hunters syndrom, men spaltelampeundersøkelse kan avsløre diskrete hornhinneskader som ikke påvirker synet. Retinal dysfunksjon kan oppdages, mens glaukom ikke er et vanlig funn.
tidlig psykomotorisk utvikling oppdages vanligvis som normalt, men i MPS I er utviklingsforsinkelsen vanligvis åpenbar ved 18 måneders alder, med en progressiv mental tilbakegang som fører til alvorlig intellektuell funksjonshemning. Språkkunnskaper er svært begrenset i disse barna. En global forsinkelse av utviklingsmessige milepæler i MPS II blir tydelig fra 2 år fremover . Det ledende nevrologiske trekket ved alvorlig Huntersykdom er imidlertid representert ved markerte atferdsforstyrrelser, som hyperaktivitet, obstinacy og aggressivitet, som vanligvis ikke observeres hos de med svekket fenotype .
Mukopolysakkaridose TYPE VII (MPS VII; OMIM #253220), også kjent som Sly syndrom, er en ultra sjelden mps karakterisert ved mangelfull aktivitet av β-glukuronidase, med lysosomal lagring av kondroitinsulfat (CS), DS OG HS, som fører til cellulær og organdysfunksjon. Den første pasienten ble beskrevet Av Sly et al. i 1973 og totalt 145 pasienter har blitt rapportert i litteraturen hittil.
den kliniske presentasjonen og sykdomsprogresjonen til MPS VII spenner over et bredt alvorlighetsgrad, fra tidlige, alvorlige, multisystem manifestasjoner og død i de aller første månedene, til en mildere fenotype med senere utbrudd, normal eller nær normal intelligens, og lengre overlevelse .
Fenotypekarakteristika hos pasienter med MPS VII ligner egenskapene TIL mps I OG MPS II (kortvoksthet, skjelettdysplasi, makrocefali, gingivalhypertrofi, tilbakevendende øreinfeksjoner, hepatosplenomegali, brokk, hjerteintervensjon, nedsatt lungefunksjon og kognitiv svekkelse). En uventet høy andel av beskrevne pasienter (41%) har en historie med ikke-immun neonatal hydrops fetalis (NIHF) . Til tross for prenatal utbruddet av sykdommen hos disse pasientene, 13 av 23 overlevde barndom med en mild til middels kurs i slutten av tenårene. TILSTEDEVÆRELSEN AV NIHF forutsier således ikke i seg selv den mulige alvorlighetsgraden av det kliniske kurset dersom pasienten overlever barndom. Et annet tidlig tegn er makrokrani, mens hjerteventil involvering og gjentatte infeksjoner i øvre og nedre luftveier vises i de første 2 årene av livet. Moderat mental retardasjon og progressivt hørselstap med taleforringelse er også tydelig med tiden.
oppsummert har de alvorlige formene FOR MPS i et veldig tidlig multisystem-utbrudd (innen 6 måneders levetid); MPS II er lik, men med senere utbrudd, OG MPS VII har en prenatal utbrudd med hyppig NIHF og tidlig alvorlig involvering av øvre og nedre luftveier.
MPS med stort sett nevrologisk og kognitiv involvering
Mukopolysakkaridoser TYPE III (MPS III, også Kjent Som Sanfilippo syndrom; Fig. 7) har en utbredt nevrologisk presentasjon. MPS III er en lysosomal lagringsforstyrrelse forårsaket av mangel på ett av de fire enzymene som er involvert i katabolismen AV HS. Fire forskjellige undertyper er kjent—MPS III TYPE A (omim #252900), TYPE B (OMIM #252920), TYPE C (OMIM #252930) og TYPE D (OMIM #252940)—hver på grunn av en annen enzymmangel: Type A, sulfamidase/sgsh gen; Type b, alfa-n-acetylglukosaminidase/NAGLU gen; Type C, alfa-glukosaminid N-acetyltransferase/HGSNAT gen; type d, n-acetilglucosamina-6-solfato sulfatasi/gns GEN). Alle fire typer (A Til D) har autosomal recessiv arv. MPS III ER den hyppigste AV MPS med en estimert prevalens mellom 0,3 og 4.1 tilfeller for hver 100.000 nyfødte, avhengig av subtype og rase .

MPS IIIA. Samme pasient ved 2,5 og 5 år. Legg merke til det forskjellige ansiktsuttrykket, som viser progresjon av nevrologisk svekkelse
Pasienter Med Sanfilippo syndrom viser progressiv kognitiv svekkelse i andre og tredje år av livet. I motsetning til FLERTALLET AV PARLAMENTSMEDLEMMER er somatiske trekk relativt mindre tydelige, OG CNS-involvering er iøynefallende. Taleutvikling forsinkelse er vanligvis det første tegnet lagt merke til av foreldrene, men de viktigste bekymringene kan være atferdsproblemer (hyperaktivitet, engstelig og aggressiv atferd, søvnforstyrrelser), etterfulgt av progressiv mental tilbakegang . Disse symptomene kan forårsake feildiagnose som atferdsforstyrrelser, oppmerksomhetsunderskudd hyperaktivitetsforstyrrelse (ADHD) og autismespektrumforstyrrelser . Epilepsi kan også være til stede.
Søvnforstyrrelser, hvor forekomsten er rapportert opp til 80-90%, er en vanlig funksjon. De består av vanskeligheter med å sovne og hyppig nattlig oppvåkning, med fullstendig reversering av dag-nattrytmen hos noen pasienter.
Alle disse manifestasjonene er svært vanskelige å håndtere og har en enorm psykologisk innvirkning på livskvaliteten for hele familien.
generelt, selv om fenotypen kan være svært lik i de fire subtypene, synes det kliniske kurset I Sanfilippo B Og C å være preget av en mindre alvorlig fenotype .
Ikke-nevrologiske symptomer er vanligvis mindre uttalt i MPS III enn i de andre MPS. Tilbakevendende øre -, nese-og halsinfeksjoner observeres i yngre alder. Tilbakevendende otitis, mellomøret ossicle defekter, og abnormiteter i det indre øret kan føre til døvhet. Et overskudd av tykke sekreter og anatomiske endringer kan gi obstruksjon av luftveiene. I de første årene av livet rapporteres diare ofte, mens forstoppelse er vanlig hos eldre pasienter.
Fysisk undersøkelse kan vise grove ansiktstrekk, selv om disse er mindre uttalt. Pasienter har makrocefali, brede øyenbryn med medial flaring og synophrys, håret er vanligvis tørt og grovt, og hypertrichose er vanlig. Hos yngre pasienter observeres mild hepatomegali og navlestreng og inguinal brokk ofte. Sjeldne, og ofte milde, kontrakturer finnes for det meste i albuene. Veksten påvirkes hos omtrent halvparten av pasientene eldre enn 12 år .
oppsummert har MPS III eller Sanfilippos sykdom en fremtredende nevrologisk involvering med svært milde somatiske tegn. Småbarn i alderen 2-3 år som utvikler hyperaktivitet, ADHD og aggressiv atferd kan mistenkes FOR Å ha MPS III.
MPS med utbredt somatisk involvering
MPS IV OG MPS VI tilstede med bare somatisk involvering. Dette er progressive forhold som sparer intellektuell evne og hovedsakelig påvirker skjelettet (MPS IVA), eller alle organer/systemer i kroppen (mps VI). Arv er autosomal recessiv. Hastigheten som symptomene forverres varierer blant berørte personer.
Mukopolysakkaridose IV (MPS IV, også kjent Som Morquio syndrom; Fig. 8) presenterer som to genetisk distinkte lidelser, hver med en annen enzymmangel: MPS Iva (Morquio a syndrom; OMIM #253000) på grunn av mangel På n-acetylgalaktosamin-6-sulfatase (GALNS gen), noe som resulterer i akkumulering av keratansulfat (KS) og CS ; og MPS IVB (Morquio b syndrom; OMIM #253010) på grunn av mangel på beta-galaktosidaseaktivitet som fører til urin KS og oligosakkaridutskillelse som HOS GM1-pasienter. MPS IVB er faktisk allelisk til ULIKE former FOR GM1-gangliosidose, men mangler psykomotorisk forverring sett I GM1 gangliosidose .

MPS IVA. en 4 år gammel pasient med b antero-posterior og c lateral x-stråler som viser “padle-formet” ribbeina og flate og avrundede ryggvirvler med en typisk “anterior beaking” aspekt
Pasienter med alvorlig MPS IVA uten påviselig enzymaktivitet avslører sine første symptomer vanligvis i løpet av det første leveåret, selv om de sjelden diagnostiseres før 4-5 år. Informasjon om Naturhistorien Til Morquio a-pasienter er hovedsakelig avledet fra to omfattende samlinger av data . Redusert vekst begynner på ca 18 måneders alder og veksten vil stoppe på ca 7 eller 8 år. I motsetning Til DE andre MPS, Morquio a leddene er vanligvis lax og svært fleksibel (hypermobile), men noen ledd (vanligvis hofter og skuldre) kan ha et begrenset utvalg av bevegelse. Pasienter med slik skjelettlidelse kan få en diagnose av, eller gjennomgå evaluering for, spondyloepifyseal dysplasi, pseudoakondroplasi, multippel epifyseal dysplasi eller bilateral Legg-Calvé-Perthes sykdom .
en konstant funksjon er odontoid hypoplasi; dermed kan dislokasjon av atlanto-occipital ledd oppstå som forårsaker kompresjon og skade på bulbar og ryggmargen, noe som resulterer i lammelse eller til og med død hvis livmorhalsstabilisering ikke utføres tidlig.
MPS IVB-pasienter har en svært lik klinisk manifestasjon, men med en mindre fremtredende kort statur. De har mildt grove ansiktsegenskaper, hørselstap, hornhinneopasiteter, hjerteklaffsykdom og hyppige øvre luftveisinfeksjoner. De kan også presentere med inguinal brokk og mild hepatomegali. Skjelett dysplastiske funksjoner inkluderer platyspondyly, odontoid hypoplasia med mulig cervical subluxation, kypho-skoliose, coxa valga, og innsnevrede iliac vinger .
Mukopolysakkaridose VI (MPS VI, også kjent Som Maroteaux-Lamy syndrom; OMIM #253200; Fig. 9) skyldes patogene mutasjoner i arylsulfatase b (ARSB) genet som ligger på kromosom 5. Som en konsekvens, redusert eller fraværende aktivitet av enzymet arylsulfatase B (ASB) (n-acetylgalaktosamin 4-sulfatase) hemmer degradering AV DS bestemme sin cellulære akkumulering .

MPS VI. en 2 år gammel gutt med tydelig grov ansikt og skjelettdysostose
Pasienter uten påviselig enzymaktivitet har den alvorlige formen FOR MPS VI og viser symptomer i tidlig barndom. Deres somatiske fenotype ligner Hurler syndrom. I de fleste tilfeller, ved 2 eller 3 år, blir alvorlig og progressiv skjelettskade, kalt dysostose multiplex, tydelig. Denne skjelettdysplasi inkluderer hoftedysplasi med dysplastisk lårbenshode, unormale vertebrale legemer, uregelmessige krageben, hypoplastisk distal ulna og radius, og dysplastiske og korte metakarpale bein; carpal og tarsal bein er hypoplastiske og har en uregelmessig profil. Veksthastigheten avtar ofte etter første leveår, med en slutthøyde som regel lavere enn 120 cm. Som i andre MPS er et tidlig tegn grove ansiktsegenskaper. I tillegg har pasienter en typisk utstående underliv, hepatomegali, navlestreng og / eller inguinal brokk og hjerteinnblanding i tidlig barndom. Selv om det ikke foreligger primær utviklingshemming, kan hydrocephalus ses hos noen pasienter med påfølgende intrakraniell hypertensjon og papilledema. Pectus carinatum, stive og kontraherte ledd, skoliose og kyphos og cervikal ryggmargskompresjon forverres med tiden .
OPPSUMMERT viser MPS IV OG VI ikke CNS-involvering. MPS IV er et unikt MPS i viser felles slapphet, mens alvorlig mps VI utbruddet er lik den observert I MPS I.