artikel
Josivan Gomes Lima1*, Marcel Catão Ferreira dos Santos1, Julliane Tamara Araújo de Melo Campos2
1Departamento de medicina clínica, disciplina de endocrinologia e metabologia. Hospital Universitário Onofre Lopes, Universidade Federal do Rio Grande do Norte (UFRN), Natal, RN, Brazilië
2Faculty of Health Sciences of Trairi, Federal University Of Rio Grande do North (UFRN), Natal, RN, Brazilië
Abstract
congenitale gegeneraliseerde lipodystrofie (CGL) is een zeldzame en ernstige autosomaal recessieve ziekte. Patiënten zijn gebrekkig in de opslag van lichaamsvet en daarom leggen ze vet af in ectopische weefsels, voornamelijk lever, en kunnen cirrose ontwikkelen. Insulineresistentie is een typische bevinding, waardoor diabetes die hoge dagelijkse doses insuline vereisen. In de staat Rio Grande do Norte, Brazilië, hebben we een van de grootste cohorten van patiënten met CGL. In dit artikel beoordelen we pathofysiologie, klinisch beeld en behandeling van deze ziekte.
introductie
type 2 diabetes is een wereldgezondheidsprobleem en is gewoonlijk het gevolg van overmatig gewicht en toegenomen visceraal vet, wat perifere insulineresistentie veroorzaakt en het onvermogen van de alvleesklier om insuline af te geven om deze resistentie te compenseren. Andere, minder vaak voorkomende vormen van diabetes komen voor als gevolg van specifieke genetische mutaties, zoals de congenitale gegeneraliseerde lipodystrofie (CGL), ook bekend als Berardinelli-Seip congenitale lipodystrofie (BSCL). CGL is een autosomaal recessieve ziekte die is geclassificeerd in vier types, gebaseerd op genmutatie. De veranderde genen spelen essentiële functies voor de vorming van adipocyte, lipideproductie en juiste opslag binnen adipocyte. De mutaties verminderen vetweefsel met de daaruit voortvloeiende afzetting van vet in ectopische plaatsen, wat leidt tot vette lever, veranderd koolhydraatmetabolisme, ernstige insulineresistentie met hyperinsulinemie en acromegaloã de kenmerken en dyslipidemia1-3. Het CGL-syndroom heeft ongeveer 500 gevallen gemeld in de wereld. In Brazilië, in de staat Rio Grande do Norte (RN), hebben we 54 gevallen gediagnosticeerd, behandeld en gevolgd in de afgelopen 20 jaar4, 5. In een beschrijvende studie met secundaire gegevens schatten we in totaal 103 patiënten in RN6. Dit wijst op een veel hogere prevalentie dan in de literatuur (1: 1 miljoen) 7.
triacylglycerolvorming en opslag in lipidendruppels
de biosynthese van triglyceriden en fosfolipiden (figuur 1A) begint met glycerol-3-fosfaat acyltransferase (GPAT) acylering van glycerol-3-fosfaat in positie 1, vorming van 1-Acylglycerol-3-fosfaat (lysofosfatidinezuur). Het wordt gevolgd door een andere acyleringsstap op positie twee door het enzym AGPAT (1-Acylglycerol-3-fosfaat acyltransferase), afkomstig van 1,2-Diacylglycerol-3-fosfaat (fosfatidinezuur). Het is een belangrijke tussenstap in de biosyntheseweg van zowel triglyceriden als fosfoglyceriden. Er zijn 11 isovormen van agpat enzymen, gecodeerd door verschillende genen4. AGPAT1 en AGPAT2 zijn het meest uitgebreid bestudeerd. AGPAT1 is in hoge mate aanwezig in testis, alvleesklier en, in mindere mate, in vetweefsel en andere weefsels zoals hart, placenta, hersenen, long, terwijl AGPAT2 overvloedig aanwezig is in vetweefsel. In de volgende stappen, ontstaat het cytosolic enzym phosphatidic zure phosphatase (PAP of lipin) 1,2-diacylglycerol, en de 1,2-diacylglycerol acyltransferase (DGAT) vormt triacylglycerol4. Fosfatidinezuur en diacylglycerol kunnen ook andere fosfolipiden zoals cardiolipin, phosphatidylinositol, en phosphatidylcholine veroorzaken.
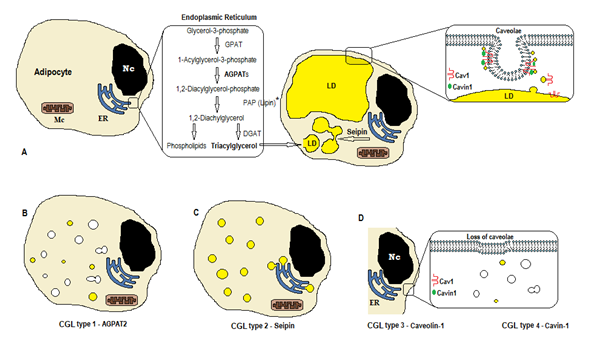
figuur 1. Schema van triglyceridensynthese volgens CGL-typen. A) normale synthese en opslag van triacylglycerol (TAG) in de adipocyt. (B) mutatie van AGPAT2 vermindert de productie van tags (sommige worden nog steeds gesynthetiseerd onder stimulatie van andere AGPATs). (C) mutatie van SEIPIN gen afname TAG synthese en lipide druppel (LD) vorming en fusie. (D) Caveoline-1 en Cavin-1 zijn nodig voor de vorming en stabilisatie van de caveolae. Mutatie in CAV1 (type 3) of CAVIN1 (type 4) kan verlies van caveolae in het membraan veroorzaken. Nc, nucleus. Endoplasmatisch reticulum. Mc, mitochondria. * Lipin is een cytosolisch enzym dat door seipin in de ER wordt verankerd.
deze reacties komen voor in het endoplasmatisch reticulum (ER) van de adipocyten, waar een progressieve accumulatie van triglyceriden de vorming van kleine lipidedruppeltjes (LD)veroorzaakt 8. Het product van het gen BSCL2 is een transmembraaneiwit genaamd seipin dat de fusie van kleine LD veroorzaakt, die grote LD voortbrengt. Seipin bevindt zich in de ER en concentreert zich op de kruising met ontluikende LD, waardoor het lipidenverkeer tussen ER en LD en de integratie van triglyceriden in LD9 wordt vergemakkelijkt. Seipin kan ook dienst doen als anker ER aan het cytosolic enzym lipin 1. Naast noodzakelijk voor de fusie van de lipidedruppel, grootte, en morfologie, is seipin ook essentieel voor adipogenesis (via interactie aan lipin 1) en cellulaire triglyceridelipolysis10, 11. De deficiëntie van seipin belemmert de differentiatie van pre-adipocytes aan adipocytes en beà nvloedt de definitieve maturation9, zoals door studies in mesenchymal stamcellen met uitgeslagen bscl2 wordt getoond 12. De niet-vetweefsels drukken ook seipin uit, en andere functies moeten worden bepaald.
in de adipocyten zijn caveolae, die gespecialiseerde 50-100nm membraaninvaginaties zijn, goed voor 20% van het plasmamembraan gebied, waardoor de adipocyten de cellen zijn met de hoogste dichtheid van caveolae13. De vorming van lipidedruppels heeft een membraaneiwit nodig (Caveoline – de belangrijkste component van caveolae-membranen) en een cytoplasmatisch eiwit (Cavine-1)14. De genen CAV1, CAV2 en CAV3 coderen drie vormen van caveoline met vergelijkbare structuren (respectievelijk Caveoline-1, Caveoline-2 en Caveoline-3). Caveolin-1 en Caveolin-2 zijn aanwezig in adipocytes, fibroblast, en endothelial cellen, en Caveolin-3 is aanwezig slechts in skeletachtige en hartspier13, 15. Caveolin-1 is de belangrijkste en meest bestudeerde. Het wordt uitgedrukt in twee verschillende isovormen (1a en 1b). Caveolin-1 transloceert van het plasmamembraan naar lipidendrupplet, hetgeen noodzakelijk is voor lipidenhandel en metabolisme16. Lipidedruppeltjes slaan triglyceriden op na het voeden en deze molecules worden gehydrolyseerd tot vetzuur, en vrijgegeven tijdens het vasten; dit mechanisme kan door Caveolin-116 worden geregeld. Caveolin-1-deficiëntie verhoogt ook de gevoeligheid voor celdood door autofagie17.
het gen CAVIN1 codeert voor een cytoplasmatisch eiwit genaamd caveolae associated protein 1 (Cavin-1)14, 16, dat verplicht is voor de vorming en stabilisatie van caveolae. Cavin-1 wordt uitgedrukt in adipocytes, spiercellen, en andere cellen, en is ook essentieel in de transmissie van caveolae-voortgekomen signalen14, 18. Knockout van het CAV1 gen veroorzaakt een gebrek aan caveolae in niet-spiercellen, terwijl de knockout van CAVIN1 de afwezigheid van caveolae in alle weefsels veroorzaakt, met inbegrip van muscle14. Het gebrek aan caveolae kan de regulering van lipolyse, vetzuurflux, triglyceridesynthese, en de signalen van andere wegen beà nvloeden.
typen CGL
op basis van detecteerbare genetische veranderingen worden vier typen beschreven. Type 1 en 2 zijn verantwoordelijk voor meer dan 95% van de gevallen, en type 2 heeft een ernstiger getroffen fenotype. Slechts één geval van type 3 en ongeveer 30 gevallen van type 4 zijn gemeld.4
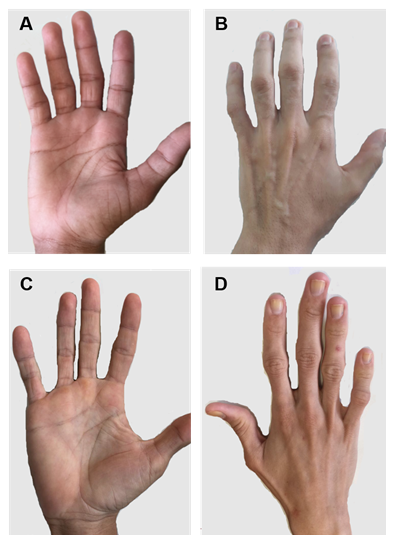
Figuur 2. Handen van patiënten met CGL type 1 en 2. A) en B) anterieure en posterieure weergave van handen van type 1-patiënten. Blijkbaar normale handen, want er is nog steeds mechanisch vetweefsel. (C) en (D) anterieure en posterieure weergave van handen van type 2-patiënten. De ernst van de ziekte is groter en het gebrek aan vet is duidelijk en gemakkelijk merkbaar.
CGL type 1. In 1999, Garg et al. beschreven patiënten mutatie op chromosoom 9q34, en drie jaar later Agarwal et al. toonde AGPAT2 als het enzym beïnvloed door deze mutatie2, 19. Door mutatie van deze AGPAT2 vindt geen of minimale productie van triacylglycerol plaats door de stimulus van andere isovormen. Het fenotype van de knockoutmuizen van AGPAT2 is vergelijkbaar met dat van mensen met het CGL-type, wat de rol van dit enzym in de pathofysiologie20, 21 bevestigt.
CGL Type 2. Magre et al. waren de eersten die de mutatie in het seipin-gen (chromosoom 11q13)identificeerden 3. Mutaties (meestal onzin) van het seipin-gen (BSCL2) produceren een afgekapt eiwit en kunnen het lipidenmetabolisme beïnvloeden door verschillende mechanismen: a) vermindering van de seipinestabiliteit; B) vermindering van het vermogen om lipine 1 te binden; en C) het niet oligomeriseren en zich uitsluitend lokaliseren naar het ER-membraan11. Sommige cellen kunnen nog triacylglycerol en kleine lipidedruppeltjes produceren, maar de grote lipidedruppeltjes zijn afwezig wegens verlies van de capaciteit van fusie van deze kleine lipidedruppeltjes. Er is ook een mislukking in de uitdrukking van adipogenic factoren, zoals peroxisome proliferator-geactiveerde receptor gamma (PPARG), evenals adiponectin en adipocyte vetzuur bindende proteã ne (FABP4)11, 16. Seipinedeficiëntie tast de adipogenese aan, verhoogt de lipolyse en voorkomt triglyceridenaccumulatie in adipocyten.
CGL type 3. Dit type werd onlangs beschreven bij een patiënt die ondanks het CGL-fenotype geen mutaties in genen AGPAT2 of BSCL222 had. De muizen met een verandering in Cav1 zijn bestand tegen dieet-veroorzaakte zwaarlijvigheid en hebben insulineresistentie, hypertriglyceridemia, verminderde adiponectin, verminderde vette massa, en kleine adipocytes16. Na het kiezen van kandidaatgenen op basis van studies bij muizen, Kim et al. bevestigde de aanwezigheid van een nonsense mutatie in het caveolin-1 gen (CAV1), op chromosoom 7q3122.
CGL type 4. In dit is een zeldzaam type het aangetaste gen is de CAVIN1, die codeert voor het eiwit Cavin-1. Bij de mens is het gemeld bij patiënten met gegeneraliseerde congenitale lipodystrofie en spierdystrofie15, 23.
onlangs zijn mutaties in de pcyt1a-en PPARG-genen beschreven die lipodystrofy24, 25 veroorzaken.
klinische kenmerken
CGL patiënten vertonen gewoonlijk acromegaloid facies, acanthosis nigricans, phebomegalie, hepatomegalie en spierhypertrofy5, 26, 27. Verschillende auteurs noemen hernia navelstreng als een klinische bevinding van het syndrome26. We evalueerden de frequentie van het in onze reeks van patiënten, en geen van hen presenteerde deze verandering 28. In feite, de afwezigheid van periumbilical vetweefsel veroorzaakt uitsteeksel van de navel litteken, en dit kan ten onrechte worden gediagnosticeerd als een hernia28, 29.
zodra adipocyten vet niet voldoende kunnen opslaan, hoopt het zich op in andere weefsels, zoals de lever en spieren, wat ernstige insulineresistentie veroorzaakt. Botdensitometrie (DXA)kan een normale of hoge botmineraaldichtheid vertonen 30 en een verminderd totaal lichaamsvet (gewoonlijk lager dan 6%)27. Als gevolg van laag lichaamsvet, zijn serum adiponectin en leptin laag too27. Aangezien leptine essentieel in het controleren van honger is, hebben deze patiënten typisch hyperfagie, die gemakkelijk sinds kinderjaren duidelijk is. Adiponectin speelt een belangrijke rol aangezien een insuline sensibilisator, en zijn gebrek de insulineresistentie verergert. Desondanks zijn aanvankelijk glucose en geglyceerd hemoglobine normaal ten koste van zeer hoge insulineniveaus. Diabetes begint meestal in de puberteit; in onze serie, De gemiddelde leeftijd van begin was 15,8±7,1 jaar27. Aanvankelijk worden ze gecontroleerd met orale geneesmiddelen, die in een paar jaar hoge doses insuline nodig hebben27. Arteriële hypertensie komt voor bij een derde van de patiënten27.
er zijn enkele specifieke klinische kenmerken van elk CGL-type. Patiënten met type 1 vertonen nog steeds mechanisch vet, vooral in handpalmen, zolen, orbitale, peri-articulaire regionen31. Daarentegen vertonen type 2-patiënten een afwezigheid van metabolisch en mechanisch vetweefsel. Seipin wordt sterk uitgedrukt in de hersenen en cerebellum en is ook betrokken bij de regulering van neurale functies. Meer dan de helft van de type 2-patiënten heeft een cognitieve handicap1, 8. Type 3 en 4 hebben behoud van mechanische en beenmerg vet, en type 4 heeft spierzwakte geassocieerd met hoge serum creatine kinase en spinale instabiliteit 15.
er zijn ook genderspecifieke klinische kenmerken. Polycystische eierstokken en amenorroe komen vaak voor32. Menstruatiecycli keren gewoonlijk terug naar normaal met het gebruik van metreleptine, waarschijnlijk als gevolg van een verbetering van de insulinegevoeligheid en herstel van de LH-pulsatiliteit32. Type 2 mannen kunnen teratozoospermie hebben door het ontbreken van seipin in kiemcellen 33.
hypertriglyceridemie treedt op sinds de eerste levensjaren en kan acute pancreatitis veroorzaken. HDL is meestal lager dan 30 mg/dL. Verhogingen van leverenzymen is ook een vroege bevinding en komen uit de vetafzetting in de lever. Progressieve verlagingen van het aantal bloedplaatjes in serum wijzen op een verergering van de leverziekte en waarschijnlijke cirrose 34.
aangezien Cavin-1 aanwezig is in de spiercellen, hebben patiënten met type 4 lichte spierzwakte en verhoogde creatine kinase15.
de levensverwachting, voornamelijk bij type 2, is aanzienlijk afgenomen, waarbij overlijden niet zelden voorkwam vóór de leeftijd van 30 jaar (persoonlijke ervaring gebaseerd op 20 patiënten die in de laatste 19 jaar overleden). De oorzaken van overlijden zijn gerelateerd aan diabetes (nierfalen, plotseling overlijden), lever (cirrose, spijsverteringsbloedingen) of infecties.
diagnose en behandeling
de diagnose CGL is gebaseerd op klinische gegevens: acromegaloïde kenmerken, acanthosis nigricans, vermindering van totaal lichaamsvet, spierhypertrofie en uitsteeksel van het navelstrenglitteken. Ook kunnen laboratoriumgegevens diabetes met ernstige insulineresistentie en hypertriglyceridemie laten zien. Beeldvormingstests kunnen helpen bij het identificeren van ectopische afzettingen van vet voornamelijk in de lever en de alvleesklier (hepatische steatose met hepatomegalie en pancreassteatose). De DXA kan het lage lichaamsvet en de hoge botdichtheid bevestigen 30.
de behandeling van CGL bestaat uit een strikte beheersing van het dieet met een afname van de inname van vet, voornamelijk triglyceriden en voedingsmiddelen met een hoge glycemische index om comorbiditeiten te voorkomen en te controleeren29. Nochtans, is het ideale dieet een uitdagend doel te bereiken wegens de verhoogde eetlust en de strenge die beperking wordt bepleit. Lichaamsbeweging moet ook worden aangemoedigd om de controle van comorbiditeiten te verbeteren, behalve bij patiënten met contra-indicaties zoals ernstige cardiomyopathie29.
met betrekking tot de medicamenteuze behandeling kunnen deze patiënten behandeld worden met de gebruikelijke richtlijnen voor diabetes, hypertensie en dyslipidemie. De eerste keuze voor de behandeling van diabetes en insulineresistentie is metformine, maar meestal is het niet genoeg. In tegenstelling tot de behandeling van partiële lipodystrofie, dienen thiazolidinedionen te worden gebruikt in combinatie met caution29. Er worden andere orale antidiabetica gebruikt, maar deze werden niet specifiek onderzocht bij CGL-patiënten. Er zijn gegevens bij dieren die suggereren dat het gebruik van SGLT2-remmers (dapagliflozine) voordelen zou kunnen hebben om cardiomyopathie35 te voorkomen; studies zijn nodig om dit bij mensen te bevestigen. Naarmate de ziekte vordert en ernstige insulineresistentie optreedt, zijn hoge dagelijkse doses insuline nodig. Het gebrek aan onderhuids vetweefsel is een probleem bij het toedienen van de hoge doses insuline. Meer geconcentreerde insuline (u-300 of U500) kan nodig zijn36. Deze patiënten presenteren ernstige dyslipidemie, voornamelijk als gevolg van de toename van triglyceriden en lage HDL, en daarom is het gebruik van fibraat soms noodzakelijk om acute pancreatitis te voorkomen. Bovendien moet vanwege het hoge cardiovasculaire risico van deze patiënten interventie met een statine worden overwogen en moeten de doelen van LDL of niet-HDL strikt zijn29.
dagelijkse injecties met metreleptine veroorzaken een significante afname van de eetlust en bieden voordelen door verlaging van de glycemie, triglyceridemie en leverenzymen. Het is opmerkelijk, vooral bij kinderen, de vermindering van de buikomtrek, waarschijnlijk als gevolg van een vermindering van hepatomegalie.
conclusie
CGL is een zeldzame en ernstige ziekte die kan optreden bij diabetes (waarvoor gewoonlijk hoge doses insuline nodig zijn) en vroege sterfte. Het fenotype van de patiënt is vrij kenmerkend, waarbij echter kennis van het syndroom door de gezondheidswerkers nodig is om een vroege diagnose te stellen. Metreleptine lijkt op dit moment het enige medicijn te zijn dat de natuurlijke geschiedenis van de ziekte kan wijzigen.
belangenconflicten: geen.
- Nolis T. Exploring the pathophysiology behind the more common genetic and acquired lipodystrofies. Journal of human genetics. 2014 Jan; 59(1): 16-23.
- Agarwal AK, Arioglu E, De Almeida s, et al. AGPAT2 is gemuteerd in congenitale gegeneraliseerde lipodystrofie gekoppeld aan chromosoom 9q34. Nat Genet. 2002 mei; 31 (1): 21-3.
- Magre J, Delepine M, Khallouf E, et al. Identificatie van het gen veranderd in aangeboren lipodystrofie van Berardinelli-Seip op chromosoom 11q13. Natuurgenetica. 2001 Aug; 28 (4): 365-70.
- Patni N, Garg A. Aangeboren gegeneraliseerde lipodystrofie, nieuwe inzichten in metabole disfunctie. Natuur reviews Endocrinologie. 2015 Sep; 11 (9): 522-34.
- Garg A. verwierf en erfde lipodystrofieën. The New England journal of medicine. 2004 Mar 18; 350 (12): 1220-34.
- de Azevedo Medeiros LB, Candido Dantas VK, Craveiro Sarmento AS, et al. Hoge prevalentie van aangeboren lipodystrofie van Berardinelli-Seip in de staat Rio Grande do Norte, Noordoost Brazilië. Diabetol Metab Syndr. 2017; 9: 80.
- Chiquette E, Oral EA, Garg A, et al. Het schatten van de prevalentie van gegeneraliseerde en partiële lipodystrofie: Bevindingen en uitdagingen. Diabetes, metabool syndroom en obesitas: doelen en therapie. 2017: 375-83.
- Wee K, Yang W, Sugii S, et al. Naar een mechanistisch begrip van lipodystrofie en seipinefuncties. Biowetenschappen rapporten. 2014; 34(5).
- Dollet L, Magre J, Cariou B, et al. Functie van seipin: nieuwe inzichten uit bscl2 / seipin knockout muismodellen. Biochimie. 2014 Jan; 96: 166-72.
- Sim MF, Dennis RJ, Aubry EM, et al. De menselijke lipodystrofieproteã ne seipin is een membraanadapter ER voor adipogene pa phosphatase lipin 1. Moleculair metabolisme. 2012; 2(1): 38-46.
- Sim MF, Talukder MM, Dennis RJ, et al. Analyse van natuurlijk voorkomende mutaties in het humane lipodystrofie-eiwit seipin onthult meerdere potentiële pathogene mechanismen. Diabetologia. 2013 Nov; 56 (11): 2498-506.
- Payne VA, Grimsey N, Tuthill A, et al. Het humane lipodystrofiegen BSCL2 / seipin kan essentieel zijn voor normale adipocyte-differentiatie. Diabetes. 2008 Aug; 57 (8): 2055-60.
- Cohen AW, Hnasko R, Schubert W, et al. Rol van caveolae en caveolines in gezondheid en ziekte. Fysiologische beoordelingen. 2004 okt; 84 (4): 1341-79.
- Pilch PF, Liu L. fat caves: caveolae, lipid trafficking and lipid metabolism in adipocytes. Trends in endocrinologie en metabolisme: TEM. 2011 Aug; 22 (8): 318-24.
- Hayashi YK, Matsuda C, Ogawa M, et al. Humane ptrf-mutaties veroorzaken secundaire deficiëntie van caveolines, resulterend in spierdystrofie met gegeneraliseerde lipodystrofie. J Clin Invest. 2009 Sep; 119 (9): 2623-33.
- Parton RG, del Pozo MA. Caveolae als plasma membraansensoren, beschermers en organisatoren. Nature reviews moleculaire celbiologie. 2013 Feb; 14 (2): 98-112.
- Le Lay S, Briand N, Blouin CM, et al. Het lipoatrophic caveolin-1 deficiënte muismodel onthult autophagy in Rijpe adipocytes. Autofagie. 2010 Aug; 6( 6): 754-63.
- Liu L, Brown D, McKee m, et al. Deletie van Cavin/PTRF veroorzaakt globaal verlies van caveolae, dyslipidemie, en glucose-intolerantie. Celmetabolisme. 2008 okt; 8 (4): 310-7.
- Garg A, Wilson R, Barnes R, et al. Een gen voor aangeboren gegeneraliseerde lipodystrofie gaat over op humaan chromosoom 9q34. The Journal of clinical endocrinology and metabolism. 1999 Sep; 84 (9): 3390-4.
- Vogel P, Read R, Hansen G, et al. Pathologie van congenitale gegeneraliseerde lipodystrofie bij Agpat2 – / – muizen. Veterinaire pathologie. 2011 mei; 48 (3): 642-54.
- Cortes VA, Curtis de, Sukumaran s, et al. Moleculaire mechanismen van hepatische steatose en insulineresistentie in het agpat2-deficiënte muismodel van congenitale gegeneraliseerde lipodystrofie. Celmetabolisme. 2009 Feb; 9 (2): 165-76.
- Kim CA, Delepine M, Boutet E, et al. Associatie van een homozygote nonsense caveolin-1 mutatie met Berardinelli-Seip congenitale lipodystrofie. J Clin Endocrinol Metab. 2008 Apr; 93 (4): 1129-34.
- Rajab A, Straub V, McCann LJ, et al. Fatale hartritmestoornissen en lang-QT-syndroom in een nieuwe vorm van congenitale gegeneraliseerde lipodystrofie met spierrippling (CGL4) als gevolg van ptrf-CAVIN mutaties. PLoS genetics. 2010 Mar 12; 6 (3): e1000874.
- Payne F, Lim K, Girousse A, et al. Mutaties die de Kennedy phosphatidylcholine pathway verstoren bij mensen met aangeboren lipodystrofie en leververvetting. Proc Natl Acad Sci U S A. 2014 17 Jun; 111( 24): 8901-6.
- Dyment DA, Gibson WT, Huang L, et al. Biallelische mutaties bij PPARG veroorzaken een congenitale, gegeneraliseerde lipodystrofie vergelijkbaar met het Berardinelli-Seip-syndroom. Eur J Med Genet. 2014 Sep; 57 (9): 524-6.
- Garg A. Clinical review#: Lipodystrofieën: genetische en verworven lichaamsvetstoornissen. The Journal of clinical endocrinology and metabolism. 2011 Nov; 96 (11): 3313-25.
- Lima JG, Nobrega LH, De Lima NN, et al. Klinische en laboratoriumgegevens van een grote reeks patiënten met congenitale gegeneraliseerde lipodystrofie. Diabetol Metab Syndr. 2016; 8: 23.
- Lima GJ, Lima NN, Oliveira CF, et al. Hernia navel bij patiënten met het syndroom van Berardinelliseip: is het echt Hernia. J Clin Mol Endocrinol. 2015; 1(1): 3.
- Brown RJ, Araujo-Vilar D, Cheung PT, et al. De diagnose en het beheer van Lipodystrofiesyndromen: een Multi-Society Praktijkrichtlijn. J Clin Endocrinol Metab. 2016 Dec; 101 (12): 4500-11.
- Lima JG, Nobrega LH, Lima NN, et al. De botdichtheid bij patiënten met congenitale lipodystrofie van Berardinelli-Seip Is hoger bij trabeculaire plaatsen en bij Type 2 patiënten. J Clin Densitom. 2016 Nov 25.
- Simha V, Garg A. fenotypische heterogeniteit in de verdeling van lichaamsvet bij patiënten met congenitale gegeneraliseerde lipodystrofie veroorzaakt door mutaties in de AGPAT2-of seipin-genen. J Clin Endocrinol Metab. 2003 Nov; 88 (11): 5433-7.
- Musso C, Cochran E, Javor E, et al. Het langetermijneffect van recombinant methionyl humane leptinetherapie op hyperandrogenisme en menstruatiefunctie bij vrouwelijke en hypofyse bij mannelijke en vrouwelijke hypoleptinemische lipodystrofische patiënten. Metabolisme. 2005 Feb; 54 (2): 255-63.
- Jiang M, Gao M, Wu C, et al. Gebrek aan testiculaire seipin veroorzaakt teratozoospermie syndroom bij mannen. Proc Natl Acad Sci U S A. 2014 13 Mei; 111(19): 7054-9.
- Mitchell O, Feldman DM, Diakow M, et al. De pathofysiologie van trombocytopenie bij chronische leverziekte. Hepat Med. 2016; 8: 39-50.
- Joubert M, Jagu B, Montaigne D, et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes. 2017 Apr; 66(4): 1030-40.
- Lima JG, Lima NN, Lima RLM, et al. Glargine U300 Insulin as a Better Option than Degludec U100 to Treat a Congenital Generalized Lipodystrophy Patient. Clin Diabetes Res. 2017; 1(1).