Autosomaal recessieve congenitale ichthyosis / Actas Dermo-Sifiliográficas
Inleiding
de laatste consensusclassificatie van ichthyosis maakt onderscheid tussen twee hoofdvormen: de niet-syndromische vormen, die alleen aanwezig zijn met huidverschijnselen, en de syndromische vormen, die ook aanwezig zijn met manifestaties in andere organen (Tabel 1).1 onder de niet-syndromische vormen worden 4 groepen geïdentificeerd: gewone ichthyoses, autosomaal recessieve congenitale ichthyoses (ARCIs), keratinopathische ichthyoses en andere minder voorkomende ichthyoses.Traditioneel werd de groep van ARCIs verdeeld in 2 aandoeningen, lamellaire ichthyosis (LI) en congenitale ichthyosiforme erythroderma (CIE). In de nieuwe classificatie werd harlequin ichthyosis (HI) toegevoegd aan deze groep1 omdat inactiverende mutaties in het ABCA12 gen zijn geïdentificeerd als verantwoordelijk voor deze aandoening, 2,3 terwijl nonsense mutaties in hetzelfde gen aanleiding kunnen geven tot het LI4 of CIE5,6 fenotype. Andere minder voorkomende varianten opgenomen in de groep van ARCIs zijn self-healing collodion baby (SHCB), acral SHCB, en badpak ichthyosis.7-9
Consensusclassificatie gebaseerd op de klinische kenmerken van Ichthyosis1.
| Nonddromic Vormen | Syndromic Vormen |
| Common IchthyosesIchthyosis vulgarisRecessive x-linked ichthyosis (nonsyndromic )ajimajor formsHarlequin ichthyosisLamellar ichthyosisCongenital ichthyosiforme erythrodermaMinor formsSelf-healing collodion babyAcral self-healing collodion babyBathing pak ichthyosisKeratinopathic IchthyosesMajor formsEpidermolytic Ichthyosissuperficial epidermolytic ichthyosisminor formsannular epidermolytic ichthyosiscurth-Macklin ichthyosisautosomal recessief epidermolytic ichthyosisEpidermolytic nevusOther FormsLoricrin keratodermaErythrokeratodermia vararabilispeeling huid syndromeCongenital reticulaire ichthyosiforme erythrodermaKLICK syndroom | Syndromic X-linked Ichthyosrecessive x-linked ichthyosis (syndromic)Ichthyosis follicularis, alopecia, en fotofobie (IFAP) syndromeconradi-Scammann-happle syndroom (belletje punctata type 2)syndromic autosomaal ichthyosisskin disordersnetherton syndromeichthyosis-hypothrichosis syndromeichthyosis-Scleroserende Cholangitis syndrometrichothystrophyneurological Disorderssjögren-Larsson syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, mentale retardatie, enteropathie, doofheid, perifere neuropathie, ichthyose, keratodermie.
er zijn slechts beperkte gegevens beschikbaar over de epidemiologie van ARCIs. In de Verenigde Staten is een prevalentie bij de geboorte geschat op 1 per 100 000 inwoners voor LI en op 1 per 200 000 inwoners voor CIE. Andere studies hebben een gecombineerde prevalentie voor LI en CIE gemeld van 1 per 200 000 tot 300 000 inwoners.10,11 in sommige landen, zoals Noorwegen, is de geschatte prevalentie groter (1 op 91 000) als gevolg van mutaties in de grondlegger.12 de bevinding van 1 of meerdere terugkerende mutaties in een populatie kan zijn omdat de mutatie op een bepaald punt in de geschiedenis plaatsvond en vervolgens van generatie op generatie werd doorgegeven (mutatie van de oprichter) of omdat het gebied van het genoom waar de mutatie wordt gevonden een DNA-sequentie heeft die vatbaar is voor mutatie (mutatie hotspot). In Spanje is de geschatte prevalentie van ARCI 1 op 138 000 bij de algemene bevolking en 1 op 61 700 bij kinderen jonger dan 10 jaar.13 in bepaalde regio ‘ s van Spanje zou de prevalentie nog hoger kunnen zijn. Aan de kust van Galicië bijvoorbeeld werd een prevalentie van 1 op 33 000 gemeld, mede als gevolg van een stichter-effect.14
lamellaire Ichthyose en congenitale Ichthyosiforme Erytrodermaklinische kenmerken
hoewel aanvankelijk werd gedacht dat LI en CIE verschillende entiteiten waren, zijn er meldingen geweest van patiënten met intermediaire klinische manifestaties en beide aandoeningen kunnen worden veroorzaakt door mutaties in hetzelfde gen.Bovendien kunnen patiënten met dezelfde mutatie, zelfs binnen dezelfde familie, verschillende fenotypen ontwikkelen.12,15
de meeste patiënten worden geboren gehuld in een collodion membraan dat geleidelijk verdwijnt tijdens de eerste levensweken en wordt vervangen door het definitieve fenotype (Fig. 1 bis). Hypohidrose, ernstige warmte-intolerantie en nageldystrofie worden vaak waargenomen bij zowel LI als CIE.17-19 patiënten met LI hebben meestal ernstigere klinische manifestaties dan patiënten met CIE. Ze hebben grote platachtige schubben, vaak van een donkere kleur, die het hele lichaamsoppervlak bedekken. Erytrodermie is afwezig of minimaal. Dergelijke patiënten hebben meestal ectropion en, soms, eclabium, hypoplasie van gewrichts-en nasale kraakbeen, littekenvorming alopecia, vooral aan de rand van de hoofdhuid, en palmoplantaire keratoderma (Fig. 1 Ter en C). CIE wordt gekenmerkt door de aanwezigheid van erytrodermie en fijne witachtige schilfering (Fig. 2). Sommige patiënten hebben duidelijk erytheem en gegeneraliseerde schilfering. De schubben kunnen groot en donker gekleurd zijn, met name op de extensor oppervlakken van de benen. In minder ernstige gevallen is erytheem mild en is de schilfering prima.

klinische kenmerken van lamellaire ichthyose. Een, bruinachtige lamellaire desquamatie. B, gemerkte plantaire hyperkeratose. C, littekens alopecia van de hoofdhuid.
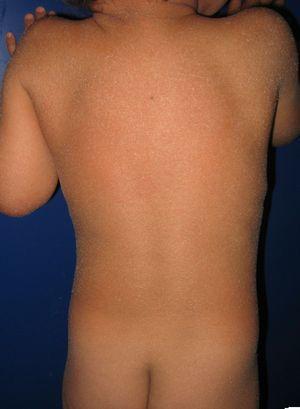
patiënt met congenitale ichthyosiforme erytrodermie en mutaties in het aloxe3 gen. Mild erytheem en gegeneraliseerde witachtige furfuraceous desquamatie kan worden gezien.
histopathologie
histopathologische veranderingen geven geen diagnose. In LI wordt massieve orthokeratotische hyperkeratose waargenomen, meestal met tweemaal de extensie zoals in CIE. De epidermis is acanthotisch en neemt af en toe een psoriasis-achtige verschijning aan. De celproliferatiesnelheid is normaal of licht verhoogd.17-19 patiënten met CIE hebben minder uitgesproken hyperkeratose, met focale of uitgebreide parakeratose, een normale of verdikte korrelige laag en meer uitgesproken acanthosis. De epidermale turnover wordt verhoogd.17-19
ultrastructuur
hoewel tot nu toe geen nauwe correlatie tussen moleculaire, klinische en ultrastructurele bevindingen is gevonden, kan elektronenmicroscopie toch nuttig zijn om andere vormen van ichthyosis uit te sluiten en in sommige gevallen als leidraad voor genetische analyses. Er zijn vier soorten aangeboren ichthyose beschreven (Tabel 2).
ultrastructurele classificatie van aangeboren Ichthyoses.
| Type | Belangrijkste Kenmerk | Overige Functies | Mutaties | Klinische Verschijnselen |
| 1 | Afwezigheid van ultrastructural markers van ichthyosis types 2, 3, en 4 | Lipide druppels of ringen in het stratum corneum (meest voorkomende)Kleine keratohyalin granulesVesicular of lobulair membraan coating korrels | TGM1 (33.3%)ALOX12B (2 gevallen) | CIE |
| 2 | Cholesterol kloven in het stratum corneum | Afwezigheid of het dunner worden van cornified envelopeSmall keratohyalin granulesLipid druppeltjes | TGM1 (89-100%) | LI |
| 3 | Gelaagd membraneous structuren in het stratum granulosum en/of stratum corneum. | Abnormale membraan coating granulesLipid dropletsFoci van prominente juxtanuclear vacuolen in de granulaire laag | NIPAL4 (93%) | CIE (meest voorkomende)LI |
| 4 | Trilamellar het membraan van de pakketten die op te vullen sommige cellen in het stratum granulosum en/of stratum corneum | Abnormale membraan coating korrels | FTAP4 | Ichthyosis prematuriteit syndroom (100%) |
Afkortingen: CIE, congenitale ichthyosiforme erythroderma; LI, lamellaire ichthyosis.
congenitale Ichthyosis type 1
congenitale ichthyosis type 1 wordt gekenmerkt door de afwezigheid van ultrastructurele markers voor ichthyosis types 2, 3 en 4. Daarom wordt de diagnose meestal alleen gesteld wanneer de andere typen zijn uitgesloten. De meest voorkomende bevinding is de aanwezigheid van lipide druppels of ringen in het stratum corneum (Fig. 3A).20 Deze lipidendruppels zijn niet een constant kenmerk of specifiek voor dit specifieke type, omdat ze niet in alle gevallen aanwezig zijn,20 en ze kunnen aanwezig zijn in andere soorten ichthyosis.Klinisch vertonen de meeste patiënten manifestaties van CIE.Een derde van de patiënten heeft mutaties in het tgm1-gen.Dit ultrastructurele type is ook geïdentificeerd in samenhang met mutaties in het alox12b gen.23,24

elektronenmicroscoop. A, congenitale ichthyosis type 1, met lipidedruppels in het stratum corneum en afwezigheid van ultrastructurele markers van de andere soorten ichthyosis. B, aangeboren ichthyosis type 2, gekenmerkt door de aanwezigheid van cholesterol spleten (pijl) in corneocyten.
congenitale Ichthyosis type 2
congenitale ichthyosis type 2 wordt gekenmerkt door cholesterolspleten in het stratum corneum (Fig. 3B).21 dergelijke spleten zijn een constante bevinding in dit type van ichthyosis, en kunnen worden gedetecteerd in verschillende biopten bij dezelfde patiënt; behandeling met orale retinoïden heeft geen invloed op deze spleten.12,25 elektrondichte aggregaten zijn ook waargenomen op corneocyten bij sommige patiënten met deficiënte tgase 1-activiteit.Klinisch vertonen de meeste patiënten ernstige manifestaties van CIE.Dit ultrastructurele type wordt sterk geassocieerd met mutaties in het tgm1 gen.12,16
congenitale Ichthyose type 3
congenitale ichthyose type 3 wordt gekenmerkt door lamellaire membraneuze structuren in het stratum granulosum en/of stratum corneum. Deze structuren zijn gerangschikt in stroken rond een lege ruimte dicht bij de kern.22,29-31 de klinische manifestaties in dit type zijn verschillend van de andere; aanvang van ichthyosis is variabel, desquamatie en erytheem kan fragmentarisch of gegeneraliseerd zijn, en de flexuren in het bijzonder worden beïnvloed. Mutaties in het nipal4 gen zijn verantwoordelijk voor 93% van de ichthyoses type 3.32
congenitale Ichthyosis Type 4
kenmerkend voor congenitale ichthyosis type 4 zijn sommige cellen in de stratum granulosum en stratum corneum gevuld met trilamellaire membraanverpakkingen.33 Deze bevindingen zijn pathognomic voor ichthyosis Prematurity syndroom, een voorwaarde momenteel beschouwd als een syndromic vorm van ichthyosis.34,35
moleculaire Studies
in genetische termen zijn de ARCIs zeer heterogeen. Het tgm1 gen is geassocieerd met de meeste gevallen, maar mutaties in 5 andere genen (ALOX12B, ALOXE3, NIPAL4, CYP4F22 en ABCA12) zijn gemeld. Fischer et al.36 bestudeerden 520 families met ARCI en identificeerden mutaties in ten minste 1 van deze genen in 78% van de gevallen (tgm1 in 32%, NIPAL4 in 16%, ALOX12B in 12%, CYP4F22 in 8%, ALOXE3 in 5% en ABCA12 in 5%). In een andere studie met 250 patiënten met ARCI van verschillende oorsprong had 38% tgm1-mutaties, 6,8% had ALOXE3-mutaties en 6,8% had ALOX12B-mutaties.37 in Galicië identificeerden we mutaties in de genen tgm1, ALOX12B, ALOXE3, NIPAL4 en CYP4F22 in 75% van de onderzochte families, maar de verdeling van mutaties was anders.14 het tgm1 gen werd gemuteerd in 68.7% van de gevallen, terwijl het aloxe3 gen bij slechts 1 patiënt werd gemuteerd. We hebben geen mutaties gevonden in een van de andere 3 onderzochte genen.
tgm1
het gen TGM1 bevindt zich op chromosoom 14q11.2 en heeft 15 exonen (GenBank NM-000359,2). Het codeert het tgase 1-enzym, een van de 3 tgase-enzymen die in de epidermis voorkomen.38 dit enzym participeert in de vorming van de cornified envelop door calcium-afhankelijke cross-linking van verscheidene proteã nen zoals involucrin, loricrin, en proline-rijke proteã nen te katalyseren.39,40 het katalyseert ook binding van ??- hydroxyceramiden in de buitenste laag van de cornified envelop met eiwitten in de binnenste laag.41,42 bij patiënten met tgm1-mutaties ontbreekt de cornified enveloppe en is de tgase 1-activiteit verminderd of niet-bestaand.43-47
sinds 1995, toen dit gen werd geïdentificeerd als verantwoordelijk voor enkele gevallen van ARCI,zijn 48-50 meer dan 110 mutaties gemeld bij patiënten van verschillende oorsprong. Mutaties in TGM1 zijn de meest voorkomende oorzaak van ARCI.36,37 deze mutatie is gevonden in 55% van de gevallen in de Verenigde Staten en in 84% van de gevallen in Noorwegen.12,51 de meest voorkomende mutatie is c.877-2A> G, die werd aangetroffen in 34% van de tot op heden gerapporteerde gemuteerde allelen.52 De hoge frequentie van deze mutatie in landen als de Verenigde Staten en Noorwegen is te wijten aan een stichtereffect.12,53 de op een na meest voorkomende mutatie is P.Arg142His. Deze en soortgelijke mutaties zijn gemeld in landen als Egypte, Duitsland, Finland en de Verenigde Staten, 15,49-51,54-56 en het lijkt erop dat dit hotspotmutaties zijn.De P. Arg307Trp mutatie komt vaak voor in de Japanse populatie.5 in Galicië, de P. Arg760X, c.1223_1227delACACA en c.984 + 1G>a mutaties in TGM1 werden geïdentificeerd in 81,82% van de families met mutaties in dit gen, wat wijst op een stichtereffect.14 bevestiging van deze hypothese werd verkregen door haplotype studie (werk nog niet gepubliceerd).
tgm1 mutaties zijn verantwoordelijk voor de meeste gevallen van LI15,27,44,46,56,58-63 en voor een klein percentage van de gevallen van CIE.43,47,64,65 dergelijke veranderingen kunnen ook tot andere vormen van ARCI zoals SHCB, acral SHCB, en badpak ichthyosis leiden.
vele studies hebben geprobeerd om genotype-fenotype associaties aan te tonen tussen mutaties in TGM1 en ultrastructurele of klinische bevindingen, maar tot op heden is geen significante correlatie waargenomen.15,16,53 over het algemeen worden patiënten met mutaties in het tgm1-gen ernstiger getroffen dan patiënten zonder dergelijke mutaties. In een studie met 83 patiënten met ARCI in Zweden en Estland werd de aanwezigheid van ectropion en collodion baby geassocieerd met tgm1 mutaties, terwijl een hoger percentage erytheem werd waargenomen bij patiënten zonder mutaties in dit gen.66 een andere studie toonde aan dat het type scaling het belangrijkste verschil is tussen dragers en niet-dragers van tgm1-mutaties, omdat werd vastgesteld dat alle patiënten met mutaties in dit gen lamellaire scaling hadden terwijl 80% van degenen zonder tgm1-mutaties fijne scaling hadden.Daarnaast is vastgesteld dat afkapmutaties vaker geassocieerd worden met hypohidrose en zweetstoornissen dan missense-mutaties.In de Noord-Amerikaanse populatie voorspelt een model gebaseerd op de aanwezigheid van bepaalde klinische kenmerken dat patiënten die geboren worden als collodion baby ‘ s en oogaandoeningen en/of alopecia hebben 4 keer meer kans hebben op tgm1 mutaties.51
aloxe3 en alox12b
de genen ALOXE3 en ALOX12B bevinden zich op chromosoom 17p13.1.67 zij hebben een vergelijkbare structuur met 15 exonen die de epidermale loxs eLOX-3 en 12R-LOX coderen.68,69 het feit dat ze voornamelijk worden uitgedrukt in de suprabasale lagen van de epidermis ondersteunt hun rol in geavanceerde fasen van epidermale differentiatie, met deelname aan de verwerking van lamellaire lichamen.24,70 deze enzymen werken op aangrenzende stappen in de hepoxilin-route (Fig. 4). 12R-LOX transformeert arachidonzuur in 12R-hydroxyeicosatetraeenzuur terwijl eLOX-3 Dit product omzet in een epoxyalcoholisomer69,71 van de Hepoxiline A3 familie.Het hepoxilineproduct is onstabiel en wordt in cellen gehydrolyseerd tot een specifiek trihydroxyderivaat (trioxiline). Hoewel de exacte rol van de producten van de hepoxilinweg niet gekend is, is er gespeculeerd dat zij aan de vorming van intercellulaire lipiden van het stratum corneum kunnen deelnemen of als signalen voor het veroorzaken van keratinocytedifferentiatie handelen.
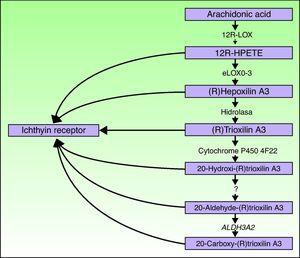
schematisch schema van de hepoxilineroute, met de participatie van de genen ALOXE3, ALOX12B, NIPAL4 en CYP4F22. De veranderingen in deze genen zijn verantwoordelijk voor sommige types van ARCI. HPETE geeft hydroperoxyeicosatetraeenzuur aan.
de alox12b-en ALOXE3-genen werden voor het eerst geïdentificeerd in 2002.73,74 sindsdien zijn meer dan 30 mutaties in de alox12b-gene23, 24,37,75-77 en ongeveer 10 in de aloxe3-gene37,74 en 75 gemeld. Deze mutaties zijn verantwoordelijk voor 14% tot 17% van ARCIs36, 37 en 72.2% van de SHCBs.23,78,79 de causatieve relatie tussen deze mutaties en het fenotype werd bevestigd door aan te tonen dat de katalytische activiteit van de epidermale LOX volledig werd afgeschaft bij patiënten met deze mutaties75,80 en door gebruik te maken van diermodellen die het ichthyosiforme fenotype reproduceerden dat bij mensen wordt gezien.81-83 beide genen zijn verantwoordelijk voor een vergelijkbaar percentage van Arci gevallen. Echter, het bereik van verschillende mutaties in het aloxe3 gen is beperkt, vanwege het overwicht van 2 mutaties, p.Arg234X en P.Pro630Leu, die lijken te corresponderen met hotspots.37,74,75
de patiënten met mutaties in de genen ALOXE3 en ALOX12B vertonen gewoonlijk een CIE-fenotype.74,75,77 de ernst van het schalen is mild of matig, en de schubben hebben een witachtige of lichtbruine kleur. Erytheem kan ook aanwezig zijn. Maar liefst 76% van de patiënten worden geboren als collodion baby ‘ s en 88% hebben zweten stoornissen.37 patiënten met mutaties in het alox12b-gen vertonen een beperktere, witachtige desquamatie in vergelijking met dragers van mutaties in het aloxe3-gen. In deze gevallen zijn de schubben bruinig en klevend. De aanwezigheid van erytheem, palmoplantaire hyperkeratosis, en accentuering van de palmoplantaire plooien worden ook geassocieerd met alox12b-mutaties.37
Ichthyin / NIPAL4
het nipal4-gen, ook bekend als het ichthyin-gen, bevindt zich op chromosoom 5q33. Het heeft 6 exons die een proteã ne met verscheidene transmembrane domeinen van onbekende functie coderen.Er is gesuggereerd dat het eiwitproduct deelneemt aan dezelfde metabolische route als LOX en kan fungeren als receptor voor trioxilinen A3 en B3 of voor andere metabolieten van de hepoxiline metabolische route.84 het zou dus betrokken zijn bij de vorming van lamellaire lichamen of in hun transport naar de extracellulaire ruimte.32 ter ondersteuning hiervan zijn 2 observaties. Ten eerste worden in 93% van de gevallen mutaties in dit gen geassocieerd met een ultra-structureel patroon van congenitale ichthyosis type 3, gekenmerkt door afwijkingen in de lamellaire lichamen en de aanwezigheid van langwerpige perinucleaire membranen in het stratum granulosum.32 ten tweede wordt NIPAL4 voornamelijk uitgedrukt in het stratum granulosum van de epidermis, waar de lamellaire lichamen aanwezig zijn.85
sinds de ontdekking van het nipal4-gen in 2004 zijn slechts 9 mutaties gemeld bij patiënten uit Mediterrane landen (Algerije, Turkije en Syrië), 84 Scandinavische landen,32 Pakistan,85 De Faeröer,32 en Zuid-Amerika.84
het klinische spectrum van patiënten met mutaties in dit gen is breed, zelfs onder leden van dezelfde familie. Tussen 3,7% 32 en 60% 84 worden geboren als collodion baby ‘ s. Wanneer het collodion-membraan verdwijnt, ontwikkelen de meeste patiënten de manifestaties van CIE, met fijne witachtige schubben op een erythemateuze basis op het gezicht en de romp en grotere, bruine schubben op de nek, billen en benen.84 Marked xerosis, gegeneraliseerde bruinachtige reticulaire hyperkeratotische plaques die lijken geaccentueerd in de huidplooien, en facial dyschromia kan aanwezig zijn.32,85 bovendien, palmoplantaire keratoderma is een frequente bevinding samen met occasionele vingercontracturen en gebogen vingernagels. Sommige studies hebben bevindingen meer typisch voor LI gemeld.De aanwezigheid van tekenen en symptomen van atopische dermatitis is gemeld bij sommige patiënten, hoewel in geen van deze gevallen mutaties in het FLG-gen werden gedetecteerd.85
CYP4F22
het gen FLJ39501 of CYP4F22 bevindt zich op chromosoom 19p13.12.86 Het heeft 12 exons87 en codeert een P450 cytochroom, familie 4, onderfamilie F, polypeptide 2, homolog van leukotrieen B4 – ω-hydroxylase (CYP4F2). De reactie die door het product van FLJ39501 in de huid en de substraten van die reactie wordt gekatalyseerd kan door analogie met zijn bekende homologen CYP4F2 en CYP4F3 worden afgeleid.Er is verondersteld dat CYP4F2 en CYP4F3 deelnemen aan de hepoxilin-weg door de omzetting van trioxiline A3 tot 20-hydroxy-(R)trioxiline A387 te katalyseren en dat het eindproduct van deze weg, 20-carboxy-trioxiline A3, een belangrijk biologisch regulerend effect in de huid kan hebben.89
tot op heden zijn slechts 8 mutaties van dit gen gemeld in 12 bloedverwantschapfamilies uit Mediterrane landen87 en in 1 familie van Israëlische oorsprong.62
In de families gerapporteerd door Lefèvre et al.De meeste patiënten hadden bij de geboorte een CIE-fenotype en dit ontwikkelde zich vervolgens tot LI. patiënten werden meestal geboren met duidelijke erytrodermie, zij het zonder een collodion membraan. Naarmate ze ouder werden, ontwikkelden ze een veralgemeende witgrijze schildering, die meer uitgesproken was in het periumbilische gebied, op de billen en op het onderste deel van het lichaam. Hyperlineariteit van de handpalmen en zolen en desquamatie op de hoofdhuid, in tijden van pityriasiform type, waren frequent.In een andere familie werden de 3 aangetaste leden geboren als collidion baby ‘ s en ontwikkelden intense erythroderma, gegeneraliseerde desquamatie en palmoplantaire keratoderma.62
ABCA12
in 2003 werd gemeld dat het ABCA12-gen verantwoordelijk was voor enkele gevallen van LI en in kaart werd gebracht op chromosoom 2q34.4 Vervolgens werd bevestigd dat mutaties in dit gen ook verantwoordelijk waren voor HI.2, 3abca12 codeert 53 exons, en behoort tot een familie van ABC-transporters, die adenosinetrifosfaat binden terwijl ook het vervoer van verscheidene molecules over het celmembraan vergemakkelijken.90 de leden van de subfamilie ABCA zijn allemaal betrokken bij het transport van lipiden.Deficiënte ABCA12-functie veroorzaakt lipidentransportstoornissen in lamellaire lichamen en leidt zo tot een afname van de intercellulaire lipidenspiegels in het stratum corneum.3 ultrastructurele studies hebben aangetoond dat ABCA12 zich bevindt in lamellaire lichamen geassocieerd met glycosylceramiden.91ABCA12 mutaties zijn in verband gebracht met stoornissen in de distributie en het transport van glycosylceramiden en met verlaagde niveaus van hydroxyceramiden, een van de belangrijkste componenten in de lipidenbarrière in de intercellulaire ruimten.3,6,92,93 de massieve hyperkeratose die bij deze patiënten optreedt, kan een compenserende respons zijn op een deficiënte lipidenbarrière.94 het kan ook te wijten zijn aan het ontbreken van desquamatie van de corneocyten,93 die kunnen worden veroorzaakt door defecten in het transport van bepaalde proteasen, zoals callicrein 5 en cathepsine D, als gevolg van stoornissen in de lamellaire lichamen.95 muriene modellen en in vitro studies suggereren dat abca12 mutaties ook een effect hebben op epidermale differentiatie.95-97
tot op heden zijn meer dan 50 mutaties gemeld in het ABCA12 gen bij patiënten met ARCI uit Afrika, Europa, Pakistan en Japan. De meest voorkomende mutaties zijn P.Val244SerfsTer28, 2, 98, 99 geà dentificeerd in Pakistaanse en Indiase populaties, en p. Asn1380Ser, 4 geà dentificeerd in Afrikaanse families. In beide gevallen, kunnen deze stichtende veranderingen zijn.
de omvang van de abca12 mutaties is gerelateerd aan het fenotype, waarbij mutaties geassocieerd zijn met volledig functieverlies wat leidt tot het HI-fenotype.2,3,98-102 bij LI en CIE daarentegen zijn de meeste mutaties missense en hebben ze een minder ernstig effect op de eiwitfunctie.4-6, 103 de mutaties die ten grondslag liggen aan het Li-fenotype lijken geconcentreerd te zijn in het eerste adenosinetrifosfaatbindingscassette-gebied.4 klinisch hebben patiënten met CIE en mutaties in het ABCA12 gen middelgrote schalen die iets groter zijn dan die gewoonlijk worden waargenomen bij patiënten met dit fenotype.Harlekijn ichthyosis
HI of Harlekijn foetus is een ernstige en meestal fatale vorm van ichthyosis. De kinderen zijn meestal prematuur met uitgebreide glanzende hyperkeratotische plaques, gescheiden door diepe scheuren, die het gehele integument bedekken en geometrische patronen vormen die doen denken aan kleding gedragen door harlekijnen, waardoor de aandoening zijn naam krijgt. Huidstrakheid leidt tot duidelijke eversion van de oogleden en lippen, rudimentaire ontwikkeling van gewrichts-en nasale kraakbeen en, af en toe, microcefalie. De kinderen hebben zelden wimpers of wenkbrauwen, hoewel het haar op de hoofdhuid kan worden behouden. De handen en voeten zijn gezwollen en oedemateus, en vaak bedekt met een handschoen-achtige laag. Ze kunnen vingercontracties hebben.
bij dergelijke patiënten is het risico op overlijden tijdens de neonatale periode zeer hoog.104 pulmonale ventilatie wordt aangetast; transepidermaal waterverlies leidt tot uitdroging, hydro-elektrische onbalans en thermische instabiliteit; en het risico van infecties wordt verhoogd. Aangezichtstrakheid en eclabium belemmeren zuigen en dus voeden, met de overeenkomstige verslechtering van de uitdroging. Pasgeborenen met deze aandoening leefden zelden langer dan een paar weken. In de afgelopen jaren zijn de kansen op overleving op lange termijn echter aanzienlijk toegenomen, voornamelijk als gevolg van de toediening van systemische retinoïden en de vooruitgang in intensieve neonatale zorg.In een recente studie overleefde 83% van de patiënten die met orale retinoïden werden behandeld, tegen 24% van de onbehandelde patiënten. De meeste sterfgevallen vonden plaats in de eerste 3 dagen van het leven, maar de behandeling werd pas daarna gestart bij veel van de overlevenden.Dit wijst erop dat veel van deze vroege sterfgevallen zouden hebben plaatsgevonden ongeacht de behandeling met retinoïden.
de kinderen die de neonatale periode overleven, ontwikkelen in het algemeen ernstige CIE.De aard en locatie van mutaties in het abca12 gen en de mate van verlies van de transporterfunctie kunnen de prognose bepalen.3.92.107 patiënten die een zekere mate van eiwitactiviteit behouden, zij het minimaal, hebben mogelijk een betere kans om te overleven. Dragers van homozygote mutaties hebben een hoger sterftecijfer.104
het belangrijkste histologische kenmerk van HI is de aanwezigheid van een extreem dikke en compacte orthokeratotische stratum corneum. De haarfollikels en zweetkanalen hebben prominente hyperkeratotic plug ‘ S107, 108 en hebben abnormale of afwezige lamellaire lichamen, lipideninsluitingen, of overblijfselen van organellen of kernen in de corneocytes, en afwezigheid van intercellulaire lipiden in de ultstructural studie.108.109 de haarfollikelen tonen een duidelijke concentratie van keratotic materiaal, die een kenmerkende eigenschap van HI is die voor prenatale diagnose wordt gebruikt.
tot op heden is het detectiepercentage van mutaties in het ABCA12-gen bij patiënten met HI bijna 100%, zodat dit een genetisch homogene aandoening lijkt te zijn.
Collodion Baby en zelfgenezende Collodion Baby
Collodion baby ‘ s worden gewoonlijk voortijdig geboren en perinatale morbiditeit en mortaliteit zijn verhoogd. Bij de geboorte wordt de pasgeborene bedekt door een glanzend aangeleerd transparant membraan dat doet denken aan cellofaanomwikkeling (Fig. 5). De baby ‘ s hebben ectropion, eclabium en hypoplasie van het neus-en gewrichtskraakbeen. Zuigen en pulmonale ventilatie kan worden gehinderd110 en transepidermaal verlies van water en het risico op infecties zijn verhoogd.110,111

Collodion-baby die zich vervolgens ontwikkelde tot een fenotype van lamellaire ichthyose.
Collodion baby is de gebruikelijke presentatie voor HI en CIE. Autosomaal dominante LI, 112, 113 Sjögren-Larsson-syndroom, 110 trichothyodystrofie,114 juveniele ziekte van Gaucher, 110 neutrale lipidenopslag-ziekte, Conradi-hünermann-Happle-syndroom, Hays-Wells-syndroom en ectodermale dysplasie115 kunnen zich soms ook voordoen als collodion-baby. Het membraan verdwijnt spontaan bij 10% tot 24% van de pasgeborenen, om plaats te maken voor een volledig normale huid.110.116 In het verleden werden deze gevallen beschreven als LI van de pasgeborene, 117 maar ze worden niet aangeduid als SHCB.118 sommige auteurs hebben de term zelfverbeterende collodion ichthyosis gesuggereerd omdat veel van deze patiënten, wanneer ze later in de kindertijd of als volwassenen opnieuw worden onderzocht, een variabele graad van anhidrose en warmte-intolerantie hebben en milde tekenen van ichthyosis, zoals xerose en fijne desquamatie, met name in de oksels en nek.78
noch optische microscopie, noch ultrastructurele onderzoeken van collodion baby zijn specifiek. Het verdient daarom de voorkeur de huidbiopsie uit te stellen totdat het definitieve fenotype zich heeft ontwikkeld.
mutaties in de genen tgm1,7,119ALOXE3,78 en ALOX12B23,78 en 79 zijn geïdentificeerd bij patiënten met SHCB. Alox12b-mutaties komen het meest voor. In een reeks van 15 Scandinavische patiënten met SHCB had 67% mutaties in het alox12b-gen, 25% in het aloxe3-gen en 8,3% in het tgm1-gen.78 veranderingen werden niet gevonden in sommige patiënten, en zo zijn andere genen waarschijnlijk ook betrokken. Er is gespeculeerd dat deze mutaties de enzymatische activiteit in de baarmoeder verminderen, maar niet na de geboorte.7 in de baarmoeder, waar de hydrostatische druk hoog is, zet chelatie door water het gemuteerde enzym om in een inactieve Bouw. Na de geboorte, wanneer de druk afneemt, keert het enzym terug naar zijn actieve vorm en neemt de activiteit voldoende toe om een normaal of minimaal beïnvloed fenotype te behouden.7
Acrale zelfgenezende Collodion-Baby
hoewel collodion-baby het hele lichaam beïnvloedt, zijn gevallen gemeld die beperkt zijn tot de acrale regio ‘ s. In 1952, Finlay et al.120 rapporteerde een geval van collodion membraan dat alleen de handen en voeten beïnvloedde en dat een zelfgenezende cursus volgde. Onlangs, is een nieuw geval van acral SHCB gemeld in associatie met veranderingen van het gen tgm1.Het is niet bekend waarom deze laesies beperkt zijn tot acrale gebieden, hoewel factoren die geassocieerd zijn met site-afhankelijke regulatie van enzymactiviteit in werking kunnen zijn.8
Badpak Ichthyosis
Badpak ichthyosis werd voor het eerst als onafhankelijke ARCI-variant gemeld in 2005, hoewel eerder gevallen van ichthyosis met een eigenaardige verdeling waren gemeld.121-123 het is voornamelijk gedetecteerd bij patiënten van Zuid-Afrikaanse oorsprong 9, hoewel het ook is gemeld bij personen uit Europa en Mediterrane landen.124 bij de geboorte, hebben de patiënten een algemeen collodion membraan dat dan loodsen om de kenmerkende verdeling van het schalen te verlaten. De romp, proximale regio van de armen, met inbegrip van de oksels, de nek, en de hoofdhuid worden over het algemeen beïnvloed, terwijl het centrale deel van het gezicht, de ledematen, en de bijnier regio worden meestal gespaard.9 de schubben zijn groot, lamellair, en donker van kleur. Fijnere desquamatie kan optreden in de popliteale en antecubitale fossae.De handpalmen en voetzolen hebben een milde diffuse hyperkeratose, terwijl de ruggen van de handen en voeten geen betrokkenheid vertonen.Histopathologisch onderzoek van de aangetaste huid toont uitgesproken hyperkeratose zonder parakeratose, normale korrelige lagen, lichte of matige acanthose en een mild lymfocytair infiltraat in de bovenste dermis.9 elektronenmicroscopie observaties komen in de meeste gevallen overeen met congenitale ichthyosis type 2. Onaangetaste huid toont geen abnormale bevindingen.124,125 in gezonde huid, is de activiteit van TGase 1 lichtjes verminderd en gewoonlijk gelokaliseerd in pericellular gebieden. In de betrokken huid, enzymatische activiteit is residueel en abnormaal gelegen in het cytoplasma.124
mutaties zijn waargenomen in het tgm1 gen bij alle patiënten met badpak ichthyosis die tot op heden zijn onderzocht.119,124-126 de gemeenschappelijkste verandering is P.Arg315Leu, die in de meeste Zuid-Afrikaanse patiënten is geà dentificeerd en een stichtende verandering zou kunnen zijn. Oji et al.124 stelde voor dat de huidtemperatuur een rol zou kunnen spelen in de ontwikkeling van deze manifestaties. Met behulp van digitale thermografie toonden de auteurs een sterke correlatie tussen lichaamstemperatuur en desquamatie, waarbij de heetste delen van het lichaam het meest getroffen zijn. Aufenvenne et al.Bij patiënten met badpak ichthyosis vertoonde 127 een daling van de optimale temperatuur voor tgase 1 activiteit. Deze daling werd niet waargenomen bij gezonde controlepersonen of bij patiënten met gegeneraliseerde LI. Deze daling in temperatuur zou het fenotype van deze patiënten verklaren. De optimale temperatuur is 37°C Voor het normale enzym, maar 31°C voor het gemuteerde enzym.
behandeling
het primaire doel van de behandeling bij ichthyose is het elimineren van schilfering en het verminderen van xerose zonder overmatige irritatie te veroorzaken (Tabel 3). Alvorens een beslissing te nemen over de behandeling, moet rekening worden gehouden met aspecten zoals leeftijd en geslacht van de patiënt, type en ernst van de ziekte, en omvang en plaats van de laesies.128
therapeutische strategie bij autosomaal recessieve aangeboren Ichthyoses.
| therapeutische strategie voor autosomaal recessieve congenitale ichthyoses | |
| baden en mechanische verwijdering van weegschalen | baden met natriumbicarbonaat of tarwezetmeel, maïszetmeel of rijstzetmeel; mechanische verwijdering van de schalen (1 of 2 keer per dag) |
| topische behandeling (sequentieel) | ureum bevattende moisturizerskeratinolytica met propyleenglycolgecombineerde keratinolytica (propyleenglycol, α-hydroxyzuren of ureum)Keratinolytica gecombineerd met salicylzuurtopicale retinoidsine pasgeborenen en kleine kinderen, Breng een vehiculum zonder werkzame bestanddelen aan. Vermijd ureum, salicylzuur en melkzuur vanwege het risico op systemische absorptie |
| orale behandeling | orale retinoïden (acitretine of isotretinoïne)) |
| andere maatregelen | Follow-up van ectropion door de oogarts regelmatige reiniging van het buitenoor door de oor-keel-neus specialistfysiotherapie om contracturen te voorkomen.Vermijden van inspannende activiteiten in een hoge omgevingstemperatuurhydrotherapie |
baden en mechanische verwijdering van schalen
dagelijks baden wordt aanbevolen voor patiënten met ARCI om schalen en sporen van vochtinbrengende crème mechanisch te verwijderen. Dit is gemakkelijker als de patiënt 15 tot 30 minuten in water wordt ondergedompeld. Sommige auteurs raden aan om natriumbicarbonaat toe te voegen aan het bad om de keratines te denaturaliseren en het water alkalisch te maken, en zo de verwijdering van de schalen te vergemakkelijken.129 andere producten die kunnen worden toegevoegd zijn tarwezetmeel, maïszetmeel of rijstzetmeel. Badoliën zijn niet geschikt omdat ze kunnen leiden tot occlusie met daaropvolgend risico op bacteriële proliferatie en verergering van thermoregulatie.
topische behandeling
Moisturizers en topische keratolytische middelen zijn meestal de eerste therapeutische optie. Ze verbeteren de functie van de huidbarrière en vergemakkelijken desquamatie. Lichte lokale bijwerkingen, zoals voorbijgaande pruritus, irritatie of stekend gevoel kunnen optreden.
natriumchloride, ureum, vitamine-E-acetaat, glycerol en vaseline kunnen worden gebruikt als moisturizers en smeermiddelen. Bij patiënten met dikke schilfering en uitgesproken hyperkeratose kunnen 1 of meer keratolytische middelen, zoals α-hydroxyzuren (melk-en glycolzuur), 130 salicylzuur,N-acetylcysteïne, 131-133 ureum (>5%), 134 en propyleenglycol,worden toegevoegd. Modulatoren van keratinocyte differentiatie worden ook gebruikt. Deze omvatten actuele retinoïden (tretinoïne, adapaleen, tazaroteen),135,136 calcipotriol,137 en dexpanthenol.Actuele retinoïden veroorzaken vaak irritatie en kleine, zeer pijnlijke kloven.Bovendien bestaat er een risico van absorptie en teratogeniciteit bij vruchtbare vrouwen als zij te veel worden gebruikt.138 om de effectiviteit van keratolytica en moisturizers te verbeteren, kan occlusief verband worden toegepast in specifieke gebieden die ongevoelig zijn voor behandeling.139 een additief of synergetisch effect kan ook worden bereikt door 2 of meer keratolytische middelen of moisturizers te combineren.140-142 behandeling moet worden geoptimaliseerd voor elk individu, gezien de zeer variabele aard van de aandoening en de gevoeligheid van de huid en verschillen in reactie op elke behandeling. Het optimalisatieproces kan worden geholpen door de ene kant van het lichaam anders te behandelen dan de andere om vergelijkingen mogelijk te maken. Pasgeborenen en kleine kinderen moeten worden behandeld met een medium zonder werkzame stoffen, omdat de huid zeer fijn en gevoelig is en de meeste keratolytica niet worden verdragen. Bovendien is het risico van percutane absorptie van topische producten zoals ureum, salicylzuur en melkzuur groter.143-145
systemische behandeling
orale retinoïden hebben keratolytische effecten die schalen helpen elimineren en overmatige hyperkeratose voorkomen. Zowel isotretinoïne als aromatische retinoïden (acitretine en etretinaat) zijn effectief gebleken bij de behandeling van ARCIs.128.146.147 Acitretin bij een dosis van 0,5 tot 1mg / kg / d is de wijdst gebruikte drug, vooral in patiënten met LI. 148 patiënten met CIE kunnen een vollediger reactie en bij lagere dosissen hebben.
de belangrijkste bijwerkingen zijn mucocutane aandoeningen, teratogeniciteit, musculoskeletale aandoeningen en abnormaal lipidenprofiel en transaminaseverhoging.149-152 wat de teratogeniciteit betreft, dienen de geneesmiddelen in het geval van etretinaat en acitretine tijdens de zwangerschap te worden vermeden en dienen patiënten te vermijden gedurende 3 jaar na het staken van de behandeling zwanger te worden.Isotretinoïne heeft een kortere halfwaardetijd en wordt na 1 maand volledig uit het organisme geëlimineerd en kan daarom de voorkeur genieten bij vrouwen die zwanger willen worden.128
controle van de behandeling dient een laboratoriumonderzoek met een leverfunctietest en een lipidenprofiel te omvatten vóór aanvang van de behandeling, daarna 1 maand en elke 3 maanden na aanvang van de behandeling. Bij vruchtbare vrouwen moet een zwangerschapstest worden uitgevoerd in de 2 weken vóór aanvang van de behandeling en een effectieve anticonceptiemaatregel worden toegepast vanaf 4 weken vóór de behandeling tot 3 jaar daarna (in het geval van acitretin). Wanneer langdurige behandeling met retinoïden noodzakelijk is, moeten de groei en botontwikkeling worden gecontroleerd. Sommige auteurs stellen voor om vóór de behandeling een botonderzoek uit te voeren, gevolgd door een jaarlijks onderzoek.In recente richtlijnen wordt niet aanbevolen om routinematige radiografie uit te voeren vanwege de mogelijke schadelijke effecten.In plaats daarvan worden selectieve radiografische studies aanbevolen bij patiënten met atypische botpijn.152
een alternatief voor systemische retinoïdbehandeling is het gebruik van geneesmiddelen die bekend staan als retinoïnezuurmetabolismeblokkerende middelen, die de endogene retinoïnezuurspiegels verhogen. Een dergelijk medicijn is liarozol, dat de status van Wees voor de behandeling van LI, CIE en HI door het Europees Geneesmiddelenbureau en de Amerikaanse Food and Drug Administration is verleend.153-155 deze drug is getoond om efficiënter te zijn dan acitretin in klinische proeven en het wordt ook beter getolereerd en heeft een beter farmacokinetisch profiel.Andere medische zorg
bij patiënten met ectropion kan het aanbrengen van kunsttranen en oogsmeermiddelen en het hydrateren van de huid van het gezicht en de wangen in het bijzonder, het terugtrekken van de oogpalmen verminderen. Chirurgische correctie is een geldige optie in ernstige gevallen, maar dit moet meestal een paar jaar later worden herhaald. Hydrotherapie kan nuttig zijn.Patiënten dient geadviseerd te worden zware fysieke activiteit te vermijden bij hoge omgevingstemperatuur, aangezien hypohidrose het risico van een hitteberoerte en convulsies met zich meebrengt. Orale retinoïden kunnen thermoregulatie verbeteren.157 Fysiotherapie is belangrijk voor het voorkomen van flexie contractuur, met name in het geval van HI. Regelmatige reiniging van de uitwendige gehoorgang door een oor-keel-neus specialist kan voorkomen dat schubben zich ophopen en zo gehoorverlies voorkomen.
erfelijkheidsadvies en prenatale diagnose
wanneer een patiënt met ichthyose wordt gediagnosticeerd, dient hem of haar een geschikt erfelijkheidsadvies te worden aangeboden waarin de aard van de aandoening, de overdrachtsmodus en het risico op toekomstige manifestaties in de familie worden uitgelegd. Prenatale diagnose kan aangeven of de foetus is aangetast en, als dit het geval is, psychologische voorbereiding van het gezin kan worden aangeboden en problemen verwacht tijdens de zwangerschap en de geboorte. De ouders kunnen de optie van een abortus krijgen als er geen behandeling beschikbaar is. Bovendien, zou gentherapie voor deze voorwaarden in de toekomst beschikbaar komen, zou prenatale diagnose toepassing van deze therapie zo vroeg mogelijk mogelijk maken.
gedurende meer dan 20 jaar werd de prenatale diagnose uitgevoerd door een biopsiemonster van de foetale huid te nemen en deze te bestuderen met behulp van optische microscopie, elektronenmicroscopie of immunohistochemie.Deze invasieve procedure kon alleen worden uitgevoerd in de late fasen van de zwangerschap, tussen weken 15 en 23 van de zwangerschap, en werd geassocieerd met een 1% tot 3% risico op het verliezen van de foetus.160.161 de identificatie van de moleculaire mechanismen van erfelijke huidaandoeningen heeft een veel vroegere diagnose mogelijk gemaakt op basis van genetische technieken.102.162-164 foetaal DNA wordt verkregen door vruchtwaterpunctie uitgevoerd tussen week 15 en 20 of door chorionische villus bemonstering tussen week 10 en 12. Het risico op foetaal verlies met deze technieken is minder dan tussen 0,5% en 1%.165 andere niet-invasieve methodes in ontwikkeling zijn analyse van foetaal celdna en vrij foetaal DNA in maternale circulatie166 evenals het gebruik van 3-dimensionale echografie.De genetische diagnose van pre-implantatie zou ook mogelijk kunnen zijn in in-vitrofertilisatie technieken, zodat alleen bevruchte eitjes die vrij zijn van de mutatie in de baarmoeder worden geïmplanteerd, waardoor in de meeste gevallen abortus wordt vermeden.Toekomstige strategieën voor de genetische behandeling van Ichthyosis
hoewel belangrijke vooruitgang is geboekt in de genetische diagnose van ichthyosis, worden ook nieuwe strategieën voor deze ziekten nagestreefd.170 de huid is het meest toegankelijke orgaan voor gentransfertherapieën, en zo zijn dergelijke technieken minimaal invasief.171 nochtans, heeft de huid ook unieke immunologische kenmerken die geen uitdrukking op lange termijn van een transgenic product bevoordelen.In LI, slaagde een proces van ex vivo genoverdracht erin om normale tgm1 uitdrukking te herstellen en het fenotype van huid getransplanteerd op de rug van immunosuppressed muizen te corrigeren.173.174 onlangs is ook het fenotype van gekweekte keratinocyten van patiënten met HI als gevolg van mutaties in het abca12 gen teruggevonden.3
belangenconflicten
de auteurs verklaren dat zij geen belangenconflicten hebben.