De genetica van gespleten lip en gehemelte
 Intro / abstractCleft lip met of zonder gespleten gehemelte is een complexe aangeboren anomalie die geïsoleerd kan worden of samen met andere misvormingen kan worden gezien. Het kan ook deel uitmaken van het fenotype van een genetisch syndroom. Dit artikel dient als een overzicht van de prevalentie van gespleten lip en gehemelte, risico ‘s voor herhaling, en risico’ s voor andere aangeboren afwijkingen. Genetische syndromen en teratogene blootstellingen waarvan bekend is dat ze geassocieerd zijn met mondspleten zullen onderzocht worden. Daarnaast zullen genetische tests die vaak worden gevraagd in de pediatrische klinische genetica setting voor de evaluatie van de patiënt met een gespleten lip en gehemelte worden besproken.
Intro / abstractCleft lip met of zonder gespleten gehemelte is een complexe aangeboren anomalie die geïsoleerd kan worden of samen met andere misvormingen kan worden gezien. Het kan ook deel uitmaken van het fenotype van een genetisch syndroom. Dit artikel dient als een overzicht van de prevalentie van gespleten lip en gehemelte, risico ‘s voor herhaling, en risico’ s voor andere aangeboren afwijkingen. Genetische syndromen en teratogene blootstellingen waarvan bekend is dat ze geassocieerd zijn met mondspleten zullen onderzocht worden. Daarnaast zullen genetische tests die vaak worden gevraagd in de pediatrische klinische genetica setting voor de evaluatie van de patiënt met een gespleten lip en gehemelte worden besproken.
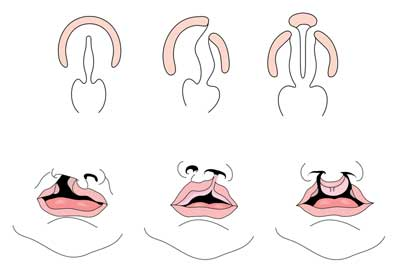
gespleten lip met of zonder gespleten gehemelte (CL/CP) verschilt van een geïsoleerd gespleten gehemelte (CP) op embryonale, epidemiologische en genetische niveaus. Gespleten lip resulteert typisch uit de maxillaire prominentie en de mediale nasale prominentie die er niet in slagen om tussen de vijfde en zesde week van embryonale ontwikkeling te smelten. De normale ontwikkeling van het gehemelte is het resultaat van de vorming van het primaire gehemelte en het secundaire gehemelte. Het primaire gehemelte wordt gevormd in weken zes tot zeven door de ontwikkeling en fusie van de mediale nasale, laterale nasale, en maxillaire processen. Het secundaire gehemelte komt voort uit de palatale planken (die zich ontwikkelen uit de gepaarde maxillaire processen van de eerste vertakkingsboog) steeds horizontaal en fuseren, de vorming van de harde en zachte gehemelte rond de negende week van embryonale ontwikkeling. Planken smelten ook met de primaire gehemelte en neustussenschot. (1)
mondspleten zijn een van de meest voorkomende geboorteafwijkingen in de neonatale kinderdagverblijf, met een algemene prevalentie van 1.6 per duizend pasgeborenen wereldwijd, met CL / CP gezien in ongeveer één per duizend geboorten en CP gezien in 0,6 per duizend geboorten. (2) Er is een hogere frequentie van CL/CP in individuen van Aziatische, Afrikaanse, en inheemse Amerikaanse afkomst. CL/CP komt ook vaker voor bij mannen. In tegenstelling, is er geen significant verschil in incidentie van CP tussen verschillende etnische achtergronden, en CP komt vaker voor bij vrouwen. (3) het risico op recidief binnen een familie hangt af van de vraag of de spleet geïsoleerd is (zonder andere klinische bevindingen) of gezien wordt als onderdeel van een genetisch syndroom. De meeste gevallen van mondspleten zijn geïsoleerd (ongeveer 80%). Geïsoleerde spleten worden verondersteld om multifactoriële overerving te hebben: ze zijn te wijten aan een combinatie van meerdere factoren, zowel genetisch als milieu. Het risico op recidief (Tabel 1) neemt toe wanneer er meer dan één betrokken familielid is. Het risico op herhaling neemt ook toe hoe ernstiger het defect is.
14sept25srrreyah gespleten lip en gehemelte kunnen worden gezien met andere aangeboren afwijkingen. De waarschijnlijkheid van een genetische of teratogene etiologie verhoogt de meer aangeboren afwijkingen waarmee een patiënt presenteert. De aanwezigheid van andere kwesties zoals intellectuele onbekwaamheid, gedragsproblemen zoals autisme, dysmorfe Eigenschappen, of andere medische zorgen zal ook een genetische wanorde of een teratogene blootstelling waarschijnlijker maken. Ongeveer 13% van de personen met een gespleten lip zal andere medische zorgen of anomalieën hebben. Het aantal stijgt tot 37% met gespleten lip en gehemelte en tot 47% met gespleten gehemelte alleen.
prenatale blootstelling aan teratogene middelen (zoals thalidomide, anticonvulsiva, alcohol, retinoïnezuur en sigaretten) en maternale aandoeningen (zoals diabetes, rodehond en folaatdeficiëntie) verhogen het risico op mondspleten. De aanwezigheid van vruchtwaterbanden verhoogt ook het risico op spleten. Het is bekend dat periconceptuele foliumzuursuppletie het risico op mondspleten vermindert.
de Pierre Robin-sequentie is een craniofaciale anomalie die wordt gekarakteriseerd door mandibulaire hypoplasie of micrognathie, secundaire U-vormige gespleten gehemelte en glossoptose die leidt tot obstructieve apneu en voedingsmoeilijkheden. De volgorde van Pierre Robin kan worden gezien als onderdeel van genetische syndromen (22q11.2 deletiesyndroom, Stickler-syndroom; hieronder beschreven). (5)
er zijn honderden genetische syndromen geassocieerd met mondspleten, waaronder cytogenetische afwijkingen (aneuploïdie, microdeleties) en enkelvoudige-gen – (Mendeliaanse) aandoeningen. Het bevestigen van een genetische diagnose is essentieel om prognose te bepalen en een risico op herhaling vast te stellen.
Aneuploïdieën zoals trisomie 13 en 18 hebben een sterke associatie met CL/CP. Trisomie 13 (aka Patau syndroom) wordt geassocieerd met drie kopieën van chromosoom 13, of onevenwichtige Robertson translocaties waarbij chromosoom 13. Baby ‘ s geboren met deze aandoening sterven meestal in de neonatale periode. Klinische kenmerken zijn onder meer gespleten lip en gehemelte, groeivertraging, ernstige malformaties van het centrale zenuwstelsel (inclusief holoprosencefalie), microcefalie, micropthalmie, iriscoloboom, afwezigheid van de ogen, misvormde oren, polydactylie, gebalde vuisten, tuimelvoeten, aangeboren hartafwijkingen en urogenitale afwijkingen. Bij trisomie 13 kunnen middellijnspleten (anders zeer zeldzaam) worden waargenomen vanwege het risico op middellijndefecten, waaronder holoprosencefalie. Trisomie 18 (aka Edwards syndroom) is typisch te wijten aan drie verschillende kopieën van chromosoom 18, en wordt geassocieerd met slechte postnatale uitkomst. Klinische kenmerken omvatten gespleten lip en gehemelte, intellectuele handicap, falen om te gedijen, aangeboren hart-en vaatziekten, hypertonie, micrognathie, kort borstbeen, lage set misvormde oren, gebalde handen, rocker bodem voeten, en hypoplastische nagels, onder anderen. Trisomie 13 en 18 kunnen gemakkelijk worden bevestigd of uitgesloten door chromosoomanalyse (karyotypering) te doen.
Microdeletiesyndromen omvatten meestal de deletie van een deel van een chromosoom. Deze schrappingen kunnen te klein zijn om door standaard karyotyping te worden ontdekt en kunnen vissen (fluorescentie in situ hybridisatie) of microarray-technologie vereisen om te worden ontdekt. Een bekend microdeletiesyndroom geassocieerd met een gespleten gehemelte is het 22q11.2-deletiesyndroom (ook bekend als Digeorge/Velocardiofaciaal syndroom). Palatale afwijkingen met inbegrip van velofaryngeale incompetentie, submucosale spleten, bifid huig, en gespleten gehemelte worden gezien in 69% van de individuen met 22q11.2 verwijdering, en kunnen deel uitmaken van de Pierre Robin sequentie. Andere klinische bevindingen zijn congenitale hartziekte, gehoorverlies, dysmorfe kenmerken, immuundeficiëntie, hypocalciëmie, nierafwijkingen, voedingsproblemen, skeletafwijkingen en psychiatrische stoornissen. Ongeveer 10% van de gevallen van 22q11. 2 deletiesyndroom wordt verondersteld familiaal te zijn. De schrapping scheidt op een autosomaal dominante manier.(6) Wolf-Hirschhorn syndroom, dat te wijten is aan een schrapping in de korte arm van chromosoom 4, wordt ook geassocieerd met mondspleten (in 25% tot 50% van de getroffen individuen). Karakteristieke gelaatstrekken (met inbegrip van prominente glabella die tot “Greek-warrior helmverschijning” leiden), aangeboren hartkwaal, intellectuele onbekwaamheid, toevallen, mislukking om te gedijen, micrognathie, preauriculaire markeringen of kuilen, en hypodontia kunnen ook als deel van de voorwaarde worden gezien.(7)
enkelvoudige genaandoeningen met mondspleten zijn onder andere het Stickler-syndroom, het Treacher Collins-syndroom en het Van der Woude-syndroom. Het Stickler-syndroom is een collageenaandoening met autosomaal dominante en, minder vaak, autosomaal recessieve overerving. Gemeenschappelijke kenmerken omvatten gespleten gehemelte (gezien als onderdeel van de Pierre Robin sequentie of zonder micrognathie), gehoorverlies (sensorineuraal en geleidend), skeletbevindingen (vroege aanvang artritis, spondyloepifysaire dysplasie), oculaire anomalieën (hoge bijziendheid, afwijkingen in het glasvocht) en karakteristieke gezichtskenmerken (met onderontwikkeling van de bovenkaak en neusbrug, middengezichtshervorming). Het genetische testen voor het syndroom van Stickler kan complex zijn, aangezien de veranderingen in ten minste zes genen in beà nvloede individuen zijn beschreven. Ongeveer 90% van de patiënten met het Stickler-syndroom heeft mutaties in het COL2A1-gen en heeft een autosomaal dominante vorm van de aandoening.(8) het syndroom van Treacher Collins is een autosomale dominante voorwaarde die door gespleten gehemelte met of zonder gespleten lip in 28% van beà nvloede individuen wordt gekenmerkt. Andere afwijkingen omvatten hypoplasie van de jukbeenderen en onderkaak, uitwendige oorafwijkingen, coloboom van het onderste ooglid, geleidend gehoorverlies, afwezigheid van lagere wimpers, preauriculaire haarverplaatsing op de wangen en choanale stenose of atresie. De diagnose van het syndroom van Treacher Collins is gebaseerd op klinische en radiografische bevindingen. Mutaties in ten minste drie genen zijn beschreven, waarbij mutaties in TCOF1 werden waargenomen bij 78% tot 93% van de patiënten.(9) het syndroom van Van der Woude wordt gekenmerkt door de aanwezigheid van congenitale, meestal bilaterale, paramediaanse onderlip fistels (pits), of soms kleine terpen met een sinuskanaal dat leidt van een slijmklier van de lip, en mondspleten (waaronder CL/CP en CP). Van der Woude is een autosomaal dominante aandoening geassocieerd met mutaties in het irf6 gen (10). Het testen voor één-gen of multi-gen voorwaarden vereist directe analyse van het gen door sequencing en/of deletie/duplicatie analyse (zoals MLPA).
aangezien genetische syndromen met gespleten lip en gehemelte geassocieerd kunnen worden met aneuploïdie, chromosoom microdeleties/microduplicaties, of enkelvoudige genaandoeningen, kan genetische testen een ingewikkeld proces zijn. Een grondige medische geschiedenis, een stamboom van drie generaties, een zwangerschapsgeschiedenis, en een dysmorfologie-examen door een klinische geneticus kan het klinische beeld verduidelijken en voor gerichte genetische tests toestaan. De nieuwere technologieën met inbegrip van microarray zullen voor de identificatie van kleine microdeletions en microduplications toestaan die eerder door standaard karyotyping worden gemist. Helaas leidt deze techniek ook tot de identificatie van verwijderingen en duplicaties van onbekende klinische betekenis, wat het genetische counseling proces compliceert. Het testen van één-gen wanorde of Mendeliaanse wanorde vereist de klinische beschikbaarheid van genetische tests voor het gewenste gen. Het kan ook duur zijn als niet gedekt door medische verzekering. Nieuwe technologieën zoals het volgende-generatie rangschikken, exome rangschikken, of genoom rangschikken (collectief bekend als genomic tests) zijn nu klinisch beschikbaar geworden. Door honderden tot duizenden genen gelijktijdig te analyseren, verhogen deze tests de diagnostische kracht en opbrengst aanzienlijk. In vergelijking met andere technieken kunnen deze tests sneller en kostenefficiënter een antwoord bieden. Op het onderzoeksgebied, heeft het exome en genoom rangschikken tot de identificatie van nieuwe genen evenals uitbreiding van de klinische eigenschappen en het spectrum voor genetische veranderingen geleid. Zoals met microarray-technologie, kunnen de genomic tests syndromen ontdekken die geen verband houden met de presentatie van de patiënt en/of reden voor het testen. Gezien de inherente complexiteit van genetische tests is geïnformeerde toestemming noodzakelijk.
conclusie
hoewel gespleten lip en gehemelte in de meeste gevallen een geïsoleerde anomalie zijn, bestaat er een sterk verband tussen mondspleten en andere anomalieën en genetische syndromen. Een genetische evaluatie door een klinische geneticus en een genetische counselor is essentieel voor anticiperende begeleiding en om risico ‘ s voor herhaling te bepalen. Genetische tests, waarvoor geïnformeerde toestemming vereist is, kunnen tijdens een genetische evaluatie worden gecoördineerd en geïnterpreteerd.Anya Revah, MS, is senior genetische counselor bij de afdeling Medische Genetica van het Maimonides Infants and Children ‘ s Hospital in Brooklyn, New York. Ze is ook een actief lid van het Maimonides Medical Center en Kings County Hospital gespleten Lip en gehemelte multidisciplinaire Team. Ze heeft een Master in de wetenschap in genetische Counseling van Boston University in Boston, Massachusetts.
1. Sadler TW. Langman ‘ s medische Embryologie. Negende Editie. Pagina 390-395.
2. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P,Mason CA, Collins JS, Kirby RS, Correa A. voor de National Birth defecten Prevention Network. Bijgewerkte Nationale schattingen van de geboorteprevalentie voor geselecteerde geboorteafwijkingen in de Verenigde Staten. 2004-2006. Onderzoek naar aangeboren afwijkingen (deel A): klinische en moleculaire Teratologie 2010; 88: 1008-1016.
3. Fraser FC. De genetica van gespleten lip en gespleten gehemelte. Is. J. Hum. Genet. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC, et al. Periconceptuele folaatinname door aanvulling en voedselinname vermindert het risico op niet-Syndrome gespleten lip met of zonder gespleten gehemelte. Prev Med 2004; 39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Ontwikkelings-en genetische perspectieven op Pierre Robin sequentie. Is. J. Med. Genet. 2013; 163C: 295-305.
6. McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11. 2 Deletiesyndroom. September. 23, 1999. . In: Pagon RA, Adam MP, Ardinger HH, et al., editor. GeneReviews . Seattle (WA): Universiteit van Washington, Seattle; 1993-2014. Beschikbaar vanaf: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia A, Carey JC, South ST, et al. Wolf-Hirschhorn Syndroom. Apr. 29, 2002. . In: Pagon RA, Adam MP, Ardinger HH, et al., editor. GeneReviews . Seattle (WA): Universiteit van Washington, Seattle; 1993-2014. Beschikbaar vanaf: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, Ala-Kokko L. Stickler syndroom. Jun. 9, 2000. . In: Pagon RA, Adam MP, Ardinger HH, et al., editor. GeneReviews . Seattle (WA): Universiteit van Washington, Seattle; 1993-2014. Beschikbaar vanaf: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs EW. Treacher Collins Syndroom. Jul. 20, 2004. . In: Pagon RA, Adam MP, Ardinger HH, et al., editor. GeneReviews . Seattle (WA): Universiteit van Washington, Seattle; 1993-2014. Beschikbaar vanaf: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy S, et al. IRF6-gerelateerde aandoeningen. Okt. 30, 2003. . In: Pagon RA, Adam MP, Ardinger HH, et al., editor. GeneReviews . Seattle (WA): Universiteit van Washington, Seattle; 1993-2014. Beschikbaar vanaf: http://www.ncbi.nlm.nih.gov/books/NBK1407/.