Frontiers in Neurologie
Inleiding
Neuroacanthocytose is een superordinaatterm voor een groep zeldzame syndromen die worden gekenmerkt door neurologische symptomen in combinatie met stekelige gedeformeerde rode bloedcellen (acanthocyten). Dit rapport richt zich op Chorea-acanthocytose (ChAc) , een weesziekte met naar schatting 1.000 aangetaste individuen wereldwijd veroorzaakt door autosomaal-recessieve mutaties in het vps13a (vacuolar protein sorting 13 homolog a) gen op chromosoom 9q (1). De ziekte loopt een progressief verloop, oorzaken van verhoogde mortaliteit kan worden afgenomen motorische functies correleren met risico voorwaarden zoals dysfagie, maar ook plotselinge onverwachte sterfgevallen zijn beschreven (2, 3). Tot nu toe is er geen oorzakelijke behandeling.
de verdachte coëxistentie van acanthocytose en bewegingsstoornissen werd voor het eerst gemeld in de jaren zeventig door Levine en Critchley (4, 5) en is sindsdien geaccepteerd als het prominente kenmerk van de ziekte. Echter, met ons casusrapport beschrijven we een zeldzaam klinisch fenotype van ChAc zonder duidelijke bewegingsstoornissen, wat een bredere verscheidenheid aan klinische presentaties suggereert. Diagnostische hulpmiddelen moeten deze uitdagingen aangaan om de juiste diagnose te stellen, stellen we hier moleculaire neuroimaging voor als een belangrijke methode.
Case Report
de man index patiënt presenteerde voor het eerst op de leeftijd van 25 jaar met twee niet uitgelokte bilaterale tonisch klonische aanvallen. Er was geen medische geschiedenis. De MRI-scan van de hersenen en EEG op die leeftijd waren normaal en er werd geen behandeling gestart vanwege de terughoudendheid van de patiënt en zeldzame voorvallen. De patiënt had echter aanhoudende aanvallen die zich nu voordoen als gedeeltelijke epilepsie. Hij beschreef terugkerende gevoelens van plotselinge duizeligheid die werden gediagnosticeerd als een duizelingwekkende aura. Verder had hij dyscognitieve aanvallen waarbij hij ofwel (A) niet reageerde en Turkse gebeden reciteerde, (b) mondelinge automatismen, of (c) rommelen met de handen en het uiten van geluiden—allemaal deels evoluerend tot tonische klonische aanvallen. Video-EEG toont nu lokale spike-Golf complexen zowel in de rechter en linker temporale kwab. In overeenstemming met de semiologie van aanvallen werd de diagnose bilaterale temporale kwabepilepsie gesteld en werd medicatie met levetiracetam gestart.
op de leeftijd van 31 aanvallen werden niet volledig onderdrukt, als onderdeel van verdere diagnostiek van temporale kwab epilepsie, onderging de patiënt een FDG-PET (figuur 1) die een bilateraal mesiotemporaal hypometabolisme vertoonde, in lijn met de mesiotemporale aanvallen oorsprong. Er was echter ook een uitgesproken, hoogst ongebruikelijk striataal hypometabolisme dat de verdenking van een neurodegeneratieve bewegingsstoornis opriep. Bijgevolg werd een uitgebreide follow-up evaluatie gestart (zie Figuur 2). Er werd een FP-CIT-SPECT uitgevoerd die een matig bilateraal verlies van de beschikbaarheid van striatale dopamine transporter opleverde, in lijn met een afname van de nigrostriatale integriteit (figuur 1). Een hoge-resolutie MRI nu gepresenteerd bilaterale atrofie van de nucleus caudatus (Figuur 3). Neurologisch onderzoek toonde een discrete gebonden gangpatroon en hypomimie, maar geen andere affectie van het extrapiramidale motorische systeem en geen bulbar symptomen zoals het voeden van dystonie. Afgezien van de lage reflexstatus van de onderste ledematen, waarvoor zenuwgeleidingsstudies een sensorimotorische axonale polyneuropathie bevestigden, was het neurologisch onderzoek normaal. Neuropsychologische symptomen omvatten aspecten van een angststoornis en de patiënt meldde moeilijkheden met kortetermijngeheugenverlies. Neuropsychologische testen bevestigden echter geen manifeste storing maar eerder een niet-specifieke afleiding, die bovendien zou kunnen zijn versterkt door onvoldoende inbeslagname onderdrukking op het moment. Laboratoriumtesten waren negatief voor symptomatische epilepsie (d.w.z. antilichamen tegen auto-immune encefalitis, antineuronale antilichamen). Cerebrospinale vloeistof vertoonde geen tekenen van ontsteking maar een matige eiwitverhoging (765 mg/l). Analyse van biogene amines in de cerebrospinale vloeistof toonde een verhoging van glutamine die we geassocieerd met recente aanvallen. In het perifere bloed werd 0,4% van de acanthocyten gevonden. Serologische testen op Wilson ‘ s ziekte waren negatief. Opvallend was een aanhoudende verhoging van creatine kinase (bereik: 1.000-3.000 U/l) die leidde tot de vermoedelijke diagnose van een mitochondriale aandoening. Voor verder onderzoek werd een ischemische lactaattest uitgevoerd die normale resultaten liet zien. Een spierbiopsie van de M. gastrocnemicus werd uitgevoerd, maar analyse van de mitochondriale functie en de ademhalingsketen was normaal.
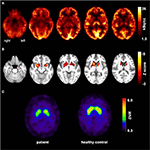
figuur 1. Resultaten van moleculaire beeldvormingsstudies. Transaxiale FDG-PET-beelden (A) toonden niet alleen een mild bilateraal mesiotemporaal hypometabolisme (in lijn met mesiotemporale insulten oorsprong) maar ook een uitgesproken striataal hypometabolisme dat zeer significant bleek te zijn in vergelijking met gezonde controles . Een extra FP-CIT-SPECT onderzoek (C) toonde ook een matig verlies van striatale dopamine transporters, getuige van een afname van nigrostriatale integriteit (een leeftijd-matched gezonde controle wordt getoond voor vergelijking; DVR, distributie volume ratio).

Figuur 2. Klinisch en diagnostisch verloop van de indexpatiënt.
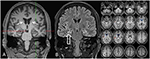
Figuur 3. MRI toont discrete bilaterale caudate head atrofie en een kleinere en hyperintense rechtszijdige hippocampus die wijst op rechtszijdige hippocampale sclerose . De linker hippocampus is enigszins hyperintense maar niet atrofisch. Bilaterale caudate head atrofie wordt bevestigd met CVR-analyse, waarbij blauwe kleuren tonen afgenomen grijze en rode kleuren verhoogd cerebrospinaal vochtvolume, respectievelijk (C).De familiegeschiedenis toonde een bloedverwantschap aan van zijn ouders (neven en nichten), die zelf geen significante medische geschiedenis hadden. Van zijn zes broers en zussen hadden er inmiddels ook twee (tussen de 20 en 30 jaar) last van algemene aanvallen (zie Figuur 4). Medische gegevens van zijn broers en zussen waren niet beschikbaar voor ons.
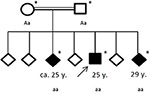
Figuur 4. Stamboom van de getroffen familie, met de leeftijd van de eerste epileptische aanval in jaren (y). Pijl: index patent. Asterisk: genetische tests uitgevoerd. Aa, heterozygoot; aa, homozygoot voor c. 4326 T>A (blz. Tyr1442*).
in de hypothese van een erfelijke neurodegeneratieve bewegingsstoornis werden de patiënt en zijn familie doorverwezen voor genetische tests. Exome sequencing onthulde een homozygote nieuwe truncerende mutatie c.4326 T>A (p.Tyr1442*) in het Chac gen VPS13A bij de drie getroffen broers en zussen. De bloedverwantschap ouders werden beiden heterozygoot ontdekt. Er werden geen andere varianten of mutaties geïdentificeerd in de exome sequencing.
in een follow-up op de leeftijd van 33 jaar had de indexpatiënt nog steeds slechts marginale extrapiramidale symptomen en geen van zijn zusters was aanwezig. Hij gaf nu de indruk van lichte stijfheid alleen onder priming op de rechter bovenste ledematen, en een milde bradykinesie van de bovenste ledematen. Tijdens de behandeling met levetiracetam en lacosamide was de patiënt vrij van aanvallen.
discussie
met dit geval tonen we aan dat ChAc zich klinisch kan manifesteren met epilepsie in de temporale kwab zonder significante motorische symptomen als gevolg van een nieuwe overeenkomstige mutatie in het vps13a-gen.
het verloop van de ziekte wordt gekenmerkt door een progressieve bewegingsstoornis (waaronder chorea, dystonie, parkinsonisme), cognitieve en psychiatrische veranderingen en myopathische symptomen met serologische markers van acanthocytose en hyperckemie. Epileptische aanvallen zijn eerder opgemerkt als een symptoom van ChAc (6) maar bewegingsstoornissen worden nog steeds beschouwd als het belangrijkste symptoom. Er zijn aanwijzingen dat epilepsie, en meer bepaald epilepsie afkomstig uit de temporale kwab, mogelijk een onderschat fenotype van ChAc is. Peluso et al. rapporteerde een patiënt die vergelijkbaar was met de Onze, die zelfs op de leeftijd van 46 jaar geen bewegingsstoornissen vertoonde terwijl hij neuroimaging-bevindingen vertoonde die wijzen op betrokkenheid van basale ganglia (7). Overeenkomstig dat, Scheid et al. beschreven drie patiënten die leden aan epilepsie en sclerose in de mesiale temporale kwab (op basis van MRI-onderzoeken) als het belangrijkste symptoom en werden gediagnosticeerd met ChAc (8). Bovendien, Mente et al. onlangs verstrekte autopsieresultaten van een ChAc-patiënt die niet alleen basale ganglia-atrofie maar ook hippocampale sclerose vertoonde (9). In lijn met ons geval lijkt bilaterale betrokkenheid van de hippocampus een gemeenschappelijke bevinding (10, 11). De exacte relatie tussen epileptische aanvallen en sclerose is echter nog steeds een kwestie van kip of ei. De correlerende rol van chorein in de structurele ontwikkeling van de hippocampus is nog niet volledig begrepen. Interessant, in een muismodel van Chac structurele verandering werd gezien, aangezien hippocampal eiwituitdrukking van GABA (A) receptor gamma 2 en zijn verankerende eiwitchorein werden verhoogd (12).
in de klinische routine kan dit fenotype nog steeds een uitdaging zijn bij het vinden van de juiste diagnose. De eerste cruciale hint naar een neurodegeneratieve ziekte bij onze patiënt was een prominente afname van het striatale glucosemetabolisme en een matige afname van de nigrostriatale integriteit op respectievelijk FDG-PET en FP-CIT-SPECT. De kleine verzameling functionele beeldvormingsresultaten, gebaseerd op de beschrijving van de casusreeksen, tonen aan dat veranderingen in het metabolisme en dopaminerge dysfunctie meestal optreden in de caudate nucleus en putamen (13). MRI van de hersenen toonde atrofie van de nucleus caudatus tijdens het verloop van de ziekte, die is beschreven als een typische bevinding in ChAc (14). Dienovereenkomstig, verschillende neurologische kenmerken omvatten meestal chorea, voeden dystonie, orofaciolinguale dyskinesieën, en tics, naast andere symptomen van basale ganglia affectie zoals parkinsonisme en dystonie (3). Het is verleidelijk om te speculeren dat de waargenomen striatale veranderingen in onze patiënt nog onder de drempel van het veroorzaken van symptomen waren (d.w.z., prodromal beeldvormende bevindingen), die uiteindelijk kunnen optreden als de ziekte vordert. Daarom moedigen we structurele en moleculaire beeldvorming aan als een gevoelig diagnostisch hulpmiddel bij deze weesziekte.
bij ChAc zijn er beschrijvingen van acanthocytenaantallen tussen 5 en 50% (3), maar er zijn enkele gevallen die aantonen dat acanthocyten pas laat in het verloop van de ziekte kunnen voorkomen (15), zeer laag kunnen zijn of zelfs volledig afwezig kunnen zijn (16). Daarom moeten acanthocyten niet als verplicht worden beschouwd voor het stellen van de diagnose. Bovendien kan epilepsie de diagnose vertragen omdat het andere typische symptomen van ChAc vermomt. Met name psychiatrische symptomen zoals persoonlijkheidsveranderingen en cognitieve achteruitgang, die zeer vaak voorkomen, kunnen verkeerd worden geïnterpreteerd en worden toegeschreven aan geneesmiddelgerelateerde bijwerkingen (d.w.z. van levetiracetam) of epileptische aanvallen. Secundaire hyperckemie wordt gezien na algemene tonische clonische aanvallen en een aanhoudende verhoging kan worden over het hoofd gezien.
ons geval benadrukt ook het cruciale voordeel van exome sequencing om een juiste diagnose te stellen, vooral bij zeldzame ziekten of wanneer de symptomen vaag zijn. De familie van zoogdiergenen vps13 (A-D) is van toenemend belang en mutaties zijn geïdentificeerd in andere neurologische ziekten zoals autosomaal recessieve ziekte van Parkinson of autosomaal recessieve spinocerebellaire ataxie (17). Recessieve mutaties in VPS13D veroorzaken bewegingsstoornissen bij kinderen (18). De onderliggende pathofysiologie van ChAc is nog niet volledig ontcijferd, maar er werd aangetoond dat VPS13A codeert voor de eiwitchoreine die alom tot expressie wordt gebracht in de hersenen en in een groot aantal andere weefsels (19). De Studies tonen aan dat het in signalerende wegen participeert die cytoskeletal architectuur, exocytose, en celoverleving (20) regelen. Een andere mutatie waarvan is beschreven dat het fenotype van een aanval gedomineerd wordt bij 9 patiënten met ChAc is de C.2343del-mutatie in het vps13a-gen (21). Het is redelijk om aan te nemen dat specifieke mutaties in hetzelfde gen resulteren in een bepaalde aberratie van chorein die een uniek fenotype veroorzaakt, bijvoorbeeld door verschillende gebieden van de hersenen te beïnvloeden. Echter, de onderliggende pathofysiologie is nog steeds alleen speculatief en verdere documentatie van de causatieve mutaties zal van belang zijn. Wij tonen hier aan dat de afkapmutatie c.4326 T>A (P.Tyr1442*), die niet eerder is beschreven, resulteert in een gelijkaardig fenotype met overheersende epilepsie bij alle aangetaste familieleden.
slotopmerkingen
epilepsie (met name bilaterale temporale kwabepilepsie) is waarschijnlijk een overheersend fenotype van ChAc. Daarom moet een zoektocht naar extra rode vlaggen (zoals hyperckemie, familiegeschiedenis, polyneuropathie) zorgvuldig worden uitgevoerd. In verdachte gevallen zou het onderzoek naar acanthocytes in het bloed en hersenen MRI moeten worden toegevoegd aangezien deze hulpmiddelen wijd beschikbaar zijn. Bij twijfel moet de diagnostiek worden aangevuld met FDG-PET en/of FP-CIT-SPECT, omdat deze gevoelig zijn bij het detecteren van striatale betrokkenheid. Bij autosomaal recessieve epilepsie met indicatieve bevindingen voor een neurodegeneratieve ziekte moet dan VPS13A single gen test of Chorein Western blot worden gebruikt om de juiste diagnose te stellen. Dit laatste moet ook worden overwogen bij andere neurodegeneratieve aandoeningen zonder genetisch gedefinieerde etiologie.
ethische verklaring
schriftelijke geïnformeerde toestemming werd verkregen van de patiënt voor deelname aan het onderzoek. Schriftelijke geïnformeerde toestemming werd verkregen van de patiënt voor de publicatie van dit rapport.
Auteursbijdragen
JW, MR en SK namen deel aan patiëntenmanagement. JW schreef de eerste versie van het manuscript. LF, HU en PM hebben cijfers opgesteld. Alle auteurs hebben bijgedragen aan de revisie van het manuscript, hebben de ingediende versie gelezen en goedgekeurd.HU is aandeelhouder van Veobrain Gmbh, een spin-off van het Universitair Medisch Centrum Freiburg.
de overige auteurs verklaren dat het onderzoek werd uitgevoerd zonder enige commerciële of financiële relatie die als een potentieel belangenconflict kon worden opgevat.
1. Walterfang M, Evans A, Leong Looi JC, Jung HH, Danek A, Walker RH, et al. De Neuropsychiatrie van neuroacanthocytose syndromen. Neurosci Biobehav Rev. (2011) 35: 1275-83. doi: 10.1016 / j.neubiorev.2011.01.001
PubMed Abstract / CrossRef Full Text / Google Scholar
2. Walker RH. Behandeling van neuroacanthocytose syndromen. Tremor Andere Hyperkinet. Mov. (2015) 5:346. doi: 10.7916 / D8W66K48
PubMed Abstract / CrossRef Full Text / Google Scholar
3. Velayos Baeza A, Dobson-Stone C, Rampoldi L, Bader B, Walker RH, Danek A, et al. Chorea-Acanthocytose. Algemene informatie. Seattle, WA: University of Washington (1993).
4. Critchley EM, Clark DB, Wikler A. Acanthocytose en neurologische aandoening zonder Betalipoproteïnemie. Arch Neurol. (1968) 18:134–40.
PubMed Abstract
5. Levine IM, Estes JW, Looney JM. Erfelijke neurologische ziekte met acanthocytose. Een nieuw syndroom. Arch Neurol. (1968) 19:403–9.
PubMed Abstract / Google Scholar
6. Hardie RJ, Pullon HW, Harding AE, Owen JS, Pires M, Daniels GL, et al. Neuroacanthocytose. een klinische, hematologische en pathologische studie van 19 gevallen. Brain(1991) 114 (Pt 1A):13-49.
PubMed Abstract / Google Scholar
7. Peluso S, Bilo L, Esposito M, Antenora A, Rosa ADR, Pappata S, et al. Chorea-acanthocytose zonder chorea: uitbreiding van het klinische fenotype. Parkinson Relat Disord. (2017) 41:124–6. doi: 10.2016 / j. parkreldis.2017.05.013
PubMed Abstract / CrossRef Full Text / Google Scholar
8. Scheid R, Bader B, Ott DV, Merkenschlager A, Danek A. ontwikkeling van mesial temporal lob epilepsie in chorea-acanthocytose. Neurologie (2009) 73: 1419-22. doi: 10.1212 / WNL.0b013e3181bd80d4
PubMed Abstract / CrossRef Full Text / Google Scholar
9. Mente K, Kim SA, Grunseich C, Hefti MM, Crary JF, Danek A, et al. Hippocampale sclerose en mesiale temporale kwab epilepsie in chorea-acanthocytose: een geval met klinische, pathologische en genetische evaluatie. Neuropathol Appl Neurobiol. (2017) 43:542–6. doi: 10.1111 / nan.12403
PubMed Abstract / CrossRef Full Text / Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert A, la Fougère C, Noachtar S, et al. Bilaterale temporale kwab epilepsie bevestigd met intracraniële EEG in chorea-acanthocytose. Seizure (2011) 20: 340-2. doi: 10.1016 / j.aanval.2010.12.007
PubMed Abstract / CrossRef Full Text / Google Scholar
11. Marson AM, Bucciantini E, Gentile E, Geda C. Neuroacanthocytose: klinische, radiologische en neurofysiologische bevindingen in een Italiaanse familie. Neurol Sci. (2003) 24:188–9. doi: 10.1007/s10072-003-0123-1
PubMed Abstract / CrossRef Full Text / Google Scholar
12. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, et al. Choreindeficiëntie leidt tot upregulatie van gefyrin en GABA(A) receptor. Biochem Biophys Res Commun. (2006) 351:438–42. doi: 10.1016/j.bbrc.2006.10.070
PubMed Abstract / CrossRef Full Text / Google Scholar
13. Ehrlich DJ, Walker RH. Functionele neuroimaging en chorea: een systematische beoordeling. J Clin Mov Disord. (2017) 4:8. doi: 10.1186 / s40734-017-0056-0
PubMed Abstract / CrossRef Full Text / Google Scholar
14. Connolly BS, Hazrati L-N, Lang AE. Neuropathologische bevindingen bij chorea-acanthocytose: nieuwe inzichten in mechanismen die ten grondslag liggen aan Parkinsonisme en epileptische aanvallen. Acta Neuropathol. (2014) 127:613–5. doi: 10.1007 / s00401-013-1241-3
PubMed Abstract / CrossRef Full Text / Google Scholar
15. Sorrentino G, De Renzo A, Miniello S, Nori O, Bonavita V. Late verschijning van acanthocytes tijdens de loop van chorea-acanthocytose. J Neurol Sci. (1999) 163:175–8.
PubMed Abstract / Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Choréo-acanthocytose sans acanthocytes. Revue Neurol. (2010) 166:100–3. doi: 10.1016 / j.neurol.2009.03.005
CrossRef Full Text / Google Scholar
17. Lesage S, Drouet V, Majoune E, Deramecourt V, Jacoupy M, Nicolas A, et al. Verlies van de vps13c-functie bij autosomaal-recessief Parkinsonisme veroorzaakt mitochondriale disfunctie en verhoogt PINK1/Parkine-afhankelijke mitofagie. Am J Hum Genet. (2016) 98:500–13. doi: 10.1016 / j. ajhg.2016.01.014
PubMed Abstract / CrossRef Full Text / Google Scholar
18. Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M, et al. Recessieve mutaties bij >VPS13D veroorzaken bewegingsstoornissen bij kinderen. Ann Neurol. (2018) 83: I6. doi: 10.1002 / ana.25204
PubMed Abstract / CrossRef Full Text / Google Scholar
19. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, et al. In vivo distributie en lokalisatie van chorein. Biochem Biophys Res Commun. (2007) 353:431–5. doi: 10.1016/j.bbrc.2006.12.059
PubMed Abstract / CrossRef Full Text / Google Scholar
20. Lang F, Pelzl L, Schöls L, Hermann A, Föller M, Schäffer te, et al. Neuronen, erytrocyten en verder – de diverse functies van chorein. Neurosignals (2017) 25: 117-26. doi: 10.1159/000485457
PubMed Abstract / CrossRef Full Text / Google Scholar
21. Benninger F, Afawi Z, Korczyn AD, Oliver KL, Pendziwiat M, Nakamura M, et al. Epileptische aanvallen als presenterend en prominent symptoom in chorea-acanthocytose met C.2343del vps13a genmutatie. Epilepsia (2016) 57: 549-56. doi: 10.1111 / epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar