Frontiers in Pediatrics
Introduction
familiale of erfelijke hartritmestoornissen omvatten significante percentages aritmieën en ook Causaal voor plotselinge hartdood (SCD) (1, 2). In de afgelopen twee decennia hebben wetenschappers en clinici enorme inspanningen geleverd om de ingewikkelde en complexe mechanismen van congenitale familiale aritmieën te ontrafelen (1-8). Om het mechanisme van aritmogenese te begrijpen, moeten we de basisprincipes van de cardiale cellulaire structuur en hun elektrofysiologische eigenschappen kennen. Hart myocytes zijn de belangrijkste werkende cellen in het hart en zij worden uitgebreid gekoppeld zodat impulsen zich snel en uniform verspreiden. Cardiomyocyten worden gescheiden van elkaar door een gespecialiseerde grens genaamd de intercalated disk; gap junction proteã nen, cardiale desmosomen, en ionenkanalen bevinden zich in de intercalated disk (9). Gap junctions bestaan uit dicht opeengepakte connexines die intercellulaire uitwisseling van kleine moleculen mogelijk maken en ook toestaan om de prikkelende stromen van een cel naar de naburige cel te stromen. Desmosomes samen met de adherens-verbindingen zijn verantwoordelijk voor de mechanische gehechtheden van individuele cardiomyocytes. Al deze componenten in de tussenliggende schijven zijn sequentieel gescheiden en elke component oefent zijn unieke functie uit; verstoring van de ene component beïnvloedt de functie van andere componenten, die het hart vatbaar maakt voor aritmieën (9-11). De cardiale ionenkanalen zijn porievormende eiwitcomplexen die voltage gated en ingewikkeld gecoördineerde binnen-en buitenbeweging van ionische stromen over de celmembranen verstrekken, essentieel voor hartritmegeneratie en propagatie. Lang QT-syndroom (LQTS), kort QT-syndroom (SQTS), sick sinus syndroom (SSS), cardiale geleidingsstoornis (CCD), BrS, catecholaminerge polymorfe ventriculaire tachycardie (CPVT), vroege repolarisatiesyndroom (ers) en familiaire atriumfibrillatie (AF) zijn de momenteel bekende cardiale kanaalopathieën, die kunnen optreden als gevolg van een enkele of meerdere defecten in de genen die verband houden met de generatie en voortplanting van het hartritme.
in dit overzicht zullen we uitsluitend LQTS-gerelateerde aritmieën, de pathofysiologie en de momenteel beschikbare klinische behandeling beschrijven. We zijn pionier in het ophelderen van genetische pathologie in familiale hartritmestoornissen in Saudi-Arabië (1, 12-14), we zijn nog steeds bezig met cardiogenetische onderzoeken in Saudi-Arabië, aan het einde van dit overzicht zullen we de momenteel beschikbare kennis bespreken over de genetische en klinische bevindingen verkregen uit verschillende Saudi-Arabische families met een geschiedenis van syncope en SCD. We zullen ook de recent gepubliceerde gegevens over LQTS toevoegen van een team van King Faisal Specialist Hospital and Research Centre, Riyad (15).
lang QT-syndroom
congenitale LQTS is een erfelijke aandoening gedefinieerd door verlenging van het QT-interval op het elektrocardiogram (ECG). Patiënten met alle vormen van LQT ‘ s zijn gepredisponeerd voor de ventriculaire tachyaritmie, torsades de pointes (TDP) wat leidt tot recidiverende syncope of SCD. In veel gevallen kan syncope of plotselinge dood de eerste en enige manifestatie zijn. LQTS treft naar schatting 1 op de 2.000 mensen wereldwijd (16). Het kenmerk van de LQTS is verlenging van het QT-interval op ECG (gecorrigeerd voor hartslag, d.w.z. QTc). Normale waarden voor QTc zijn 440 ms bij mannen en 450 ms bij vrouwen. Bij kinderen zijn leeftijdsafhankelijke en geslachtsafhankelijke waarden relevant. Recente consensus aanbeveling voor LQTS diagnose zijn de volgende (17, 18):
1. LQTS wordt gediagnosticeerd:
A. in aanwezigheid van een LQTS-risicoscore ≥3,5 in afwezigheid van een secundaire oorzaak voor QT-verlenging en/of
b. in aanwezigheid van een ondubbelzinnig pathogene mutatie in een van de LQTS-genen of
c. In de aanwezigheid van een QT-interval gecorrigeerd voor de hartslag met behulp van Bazett ‘ s formule (QTc) ≥500 ms in herhaald 12-leads elektrocardiogram en in de afwezigheid van een secundaire oorzaak voor QT-verlenging.
2. LQTS kunnen worden gediagnosticeerd in aanwezigheid van een QTc tussen 480 en 499 ms in herhaalde 12-lead ECG ‘ s bij een patiënt met onverklaarde syncope in afwezigheid van een secundaire oorzaak voor QT-verlenging en in afwezigheid van een pathogene mutatie.
patiënten met een QTc-duur ≥500 ms hadden een significant hogere cumulatieve kans op het ervaren van hun eerste syncope in vergelijking met patiënten met een QTc-duur <500 ms vanaf de geboorte tot en met de leeftijd van 20 jaar (19). Maar bij patiënten met 2, 3 en 4 syncope-episodes was het risico op een volgende syncope-episode vrijwel identiek bij patiënten met een smalle of verlengde QTc-duur (19). De moleculaire basis van LQTS is heterogeen en tot op heden zijn mutaties in 13 verschillende genen Causaal beschreven voor LQTS (1-8, 19, 20). Een genetisch defect wordt meestal gevonden in 70% van de LQTS patiënten in een van deze 13 genen (1-8, 19, 20). Onder de momenteel bekende 13 verschillende types van LQTS, de meest voorkomende zijn LQTS1, LQTS2, en LQTS3, als gevolg van defecten in cardiale ion kanaal genen, kcnq1, KCNH2, en SCN5A, respectievelijk. Negentig procent van alle LQTS causale mutaties worden gevonden in deze drie genen (1-8, 19, 20). Mutaties in de overige 10 genen zijn zeldzaam en maken slechts ∼10% uit van alle momenteel bekende LQTS mutaties.
lang QT-syndroom is gewoonlijk een autosomaal dominante ziekte, maar af en toe konden meerdere mutaties in één enkel gen of in verschillende genen worden gevonden bij 5-10% van de patiënten met LQTS (21, 22). Patiënten met meerdere mutaties kunnen een langere QTc vertonen in vergelijking met patiënten met een enkele mutatie en dergelijke patiënten hebben ook een 3,5-voudig verhoogd risico op levensbedreigende cardiale gebeurtenissen (21, 22)
Lqts1
mutaties in het kcnq1-gen zijn de meest voorkomende vorm van alle LQTS en worden LQTS type 1 (LQTS1) genoemd. Vijftig procent van alle LQTS causale mutaties worden gevonden in het kcnq1 gen (20). KvLQT1 (ook wel KV7.1 genoemd) is een eiwit dat wordt gemaakt door het kcnq1-gen en het eiwit vormt tetramers in het endoplasmatische reticulum in de cellen, die vermoedelijk co-assembleert met het eiwit nertsen (gecodeerd door KCNE1) en vervolgens worden getransporteerd naar het plasmamembraan van de cardiale myocyten, waar ze een langzaam activerende stroom bemiddelen die de repolarisatie van actiepotentieel in cardiale weefsels versnelt, deze stroom is bekend als IKs (figuur 1). Lqts1 causale kcnq1 mutaties zijn meestal missense mutaties en in zeldzame gevallen kunnen het frameshift mutaties zijn in het C-terminale gebied (23). Hartritmestoornissen in kcnq1-mutatiedragers worden veroorzaakt door adrenerge drives, bijvoorbeeld emotionele stress, fysieke inspanning, duiken, zwemmen (24-26). Patiënten met mutaties op de transmembraandomeinen van KvLQT1 lopen een hoger risico op LQTS-gerelateerde cardiale voorvallen en hebben een grotere gevoeligheid voor sympathische stimulatie (26, 27). Patiënten met missense-mutaties lopen een verhoogd risico in vergelijking met patiënten met niet-sense of truncerende mutaties (27, 28). Variatie in de 3 ‘ – UTR in het kcnq1 gen beïnvloedt ook de gevoeligheid voor aritmie significant, vermoedelijk door invloed op de expressie van het gen (29). Patiënten met aritmieën als gevolg van kcnq1-mutaties reageren vrij goed op β-blokkers, maar sommige patiënten kunnen nog steeds minder reageren of zelfs resistent zijn tegen deze medicatie. In een recent artikel, Barsheshet et al. (30) beweerde dat de patiënten met mutaties buiten de cytoplasmatische lus (C-lus) regio in de KvLQT1 minder reageren op β-blokkers.
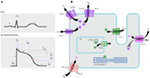
figuur 1. Ionische stromen die bijdragen tot het ventriculaire actiepotentiaal (A) en schematische weergave van een cardiomyocyt die (alleen) die eiwitten toont die betrokken zijn bij de pathogenese van erfelijke aritmiesyndromen (B). Onder A) wordt het actiepotentieel afgestemd op het geschatte tijdstip van actie tijdens het ECG. In (B) is ankyrine-B, een adapterproteïne betrokken bij het lange QT-syndroom type 4, niet afgebeeld.
homozygote of samengestelde heterozygote mutaties in het kcnq1-gen, die de recessieve vorm van de ziekte kunnen veroorzaken, het jervell-en het lange-Nielsen-syndroom (JLNS), type 1 (31), zijn vrij zeldzaam. JLNS-patiënten lijden ook aan ernstige hartritmestoornissen en doofheid (31, 32). Patiënten met kcnq1-mutaties die Causaal zijn voor JLNS hebben meestal geen functionele ik ‘ s (28, 31-33). Het is mogelijk dat sommige patiënten geen doofheid zouden kunnen hebben ondanks homozygote of samengestelde heterozygote mutaties in KCNQ1, dergelijke gevallen worden autosomaal recessieve LQTS1 (13, 32) genoemd. Bij deze patiënten kan nog een kleine hoeveelheid functionele IKs-stroom aanwezig zijn (<10% van de totale IKs), waardoor de gehoorfunctie behouden blijft, maar hun hartritmedefecten zijn even ernstig als bij JLNS-patiënten (13, 32).
LQTS2
dit type LQTS komt even vaak voor als LQTS1, goed voor 35-40% van de LQTS-patiënten met een detecteerbare mutatie (1, 20, 24, 25). KCNH2 codeert voor het Herg-eiwit (Kv11. 1), De α-subeenheid van de snel activerende vertraagde gelijkrichter K+ stroom (IKr). Pathogene mutaties in dit gen dat de kv11.1-kanaalfunctie vermindert, verlengen de duur van het QT-interval (figuur 1) en zijn Causaal voor LQTS2. Negenentwintig procent van de syncopale aanvallen in LQTS2 treden op tijdens rust / slaap en slechts 13% van de syncopale aanvallen werden gemeld tijdens inspanning (25, 34). Plotselinge verrassende geluiden, bijvoorbeeld wekker ringen, Deurbellen, telefoon gaat, meestal triggeren syncopale aanvallen bij deze patiënten (24, 34). Patiënten met mutaties in het porievormende gebied van het kv11.1 (gecodeerd door het kcnh2-gen) zijn gevoelig voor een hoog risico op hartritmestoornissen in vergelijking met patiënten met mutaties in het niet-poriegebied (35). Bij pasgeborenen wordt 2: 1 atrioventriculair (AV) blok bij voorkeur geassocieerd met kcnh2 mutaties (36). Compleet AV-blok gecompliceerd door LQTS werd ook gevonden bij 17% van de volwassen patiënten met een mutatie in het kcnh2-gen (37). Homozygote mutaties in KCNH2 zijn zeldzaam en indien aanwezig, lijden patiënten aan een ernstige vorm van LQTS, met 2:1 AV-blok en ernstige ventriculaire aritmieën, zowel tijdens intra-uteriene stadia als na de geboorte (11, 38-40). Naast de talrijke mutaties beschreven in lqts2 pathologie, kunnen gemeenschappelijke polymorfe varianten in het kcnh2 gen ook de ernst van de ziekte moduleren. Een intrigerend voorbeeld is k897t polymorfisme (SNP) in KCNH2, dat aanwezig is in 33% van de algemene bevolking (41). Er werd gemeld dat K897T de pathogeniteit van de kcnh2-mutaties verergerde (42, 43).
LQTS3
Nav1.5 is de porievormende α-subeenheid van het spanningsafhankelijke cardiale Na+ kanaal, het is een integraal membraaneiwit gecodeerd door het scn5a-gen en is betrokken bij de initiatie en geleiding van cardiale actiepotentialen. Cardiale na+ – kanalen bestaan uit een porievormende α-subeenheid (gecodeerd door SCN5A) en een of meer auxiliary β-subeenheden.
LQTS3 wordt veroorzaakt door winst van functiemutaties die de snelle inactivatie van de α-subeenheid verstoren (figuur 1) en een mutatie in het scn5a-gen is beschreven bij <10% van alle LQTS-patiënten met een mutatie (1, 20, 24). LQTS3-patiënten ervaren het merendeel (39%) van hun cardiale voorvallen tijdens slaap/rust (25), en ongeveer ∼13% van de voorvallen werd gemeld tijdens inspanning (25). In verschillende gevallen bleek één enkele scn5a-mutatie twee of zelfs drie verschillende fenotypen van aritmieën in dezelfde familie uit te oefenen, bijvoorbeeld LQTS, BrS of CCD (44-47). Mannelijke patiënten met een lqts3-mutatie zouden veel eerder symptomen kunnen ontwikkelen dan vrouwelijke patiënten (48). Zowel heterozygote als homozygote scn5a mutaties zijn beschreven in LQTS3 met functioneel 2: 1 AV blok (49). We hebben onlangs een Iraanse familie gevonden met de 1507_1509delqkp mutatie in meerdere familieleden, waar patiënten LQTS en CCD hadden gecombineerd (gegevens niet getoond). Deze mutatie is ook gemeld bij patiënten uit andere landen, wat suggereert dat dit een terugkerende en hot spot mutatie is . Mutaties met dergelijke verlies – en winst-van-functie kenmerken tijdens verschillende fasen van het actiepotentiaal werden ook gerapporteerd in 1493delK en 1795insD mutaties (44, 51).
soms kan een SNP ook een pathogeen effect hebben op zijn dragers. S1103Y is een veel voorkomende variant in het gen SCN5A, aanwezig in 13% van de Afro-Amerikanen (52). Dragers met deze variant hebben een verhoogd risico op aritmieën en wiegendood (SIDS) (52).
LQTS4
LQTS4 is de eerste niet-kanaalvorm van LQTS. Een mutatie in ANK2, een adaptereiwit, leidt tot intracellulaire calciumoverbelasting die bijdraagt aan LQTS4 (53, 54). Naast QT-verlenging kunnen patiënten met dit syndroom sinusbradycardie, paroxysmaal AF en CPVT (54) hebben. Het pathogene effect van de veranderingen ANK2 zou gematigd tot streng kunnen zijn en de klinische uitdrukkingen hangen van de strengheid van de verandering af.
LQTS5
mutaties in KCNE1 zijn geassocieerd met de Lqts5 (figuur 1) (55, 56). Heterozygote kcne1-mutaties verminderen ik ‘ s door een dominant negatief effect uit te oefenen op het bijbehorende normale allel en leiden tot vertraagde cardiale repolarisatie (figuur 1), Wat verantwoordelijk is voor een verhoogd risico op aritmieën (6). Patiënten met homozygote kcne1-mutaties lijden aan JLNS (type 2) (57, 58).
D85N is een polymorfisme in het kcne1 gen, aanwezig in 0.7-1% van de algemene bevolking (41). In een studie van Nishio et al. (59), d85n polymorfisme werd vaker gevonden in de LQTS patiënten, waardoor het een risico genotype in LQTS pathologie (potentieel alleen in de Aziatische populatie). In Europa werd D85N gemeld bij 5% van de verworven LQTS (aLQTS) patiënten (in een cohort van 32 patiënten) die TdP hadden ervaren (60).
LQTS6
kcne2 gen codeert voor MinK-gerelateerd peptide 1 (MiRP1), een vermeende β-subeenheid van het cardiale kaliumkanaal IKr (figuur 1). Mutaties in het gen KCNE2 kunnen ook leiden tot defecten in de snel activerende component van de vertraagde gelijkrichter kaliumstroom (IKr), pathologische basis van LQTS6 (61). Auditieve / akoestische stimulus zoals wekkerruis, deurbel enz. kan syncopale aanvallen veroorzaken bij kcne2-mutatiedragers, vergelijkbaar met kcnh2-mutatie (62).
LQTS7
dit syndroom is ook bekend als het syndroom van Andersen–Tawil (ATS). ATS is een zeldzame aandoening, gemanifesteerd door occasionele syncope, en hartstilstand. ECG-kenmerken omvatten lichte Qt-intervalverlenging, abnormale u-golven, frequente ventriculaire ectopie, bidirectionele ventriculaire tachycardie (VT) en polymorfe VT. Dit syndroom vertoont ook extracardiale kenmerken, bijv., skeletspier periodieke verlamming en ontwikkelingsproblemen, zoals gespleten gehemelte, lage ingestelde oren, korte gestalte, en ontwikkelingsstoornissen in de ledematen (63). Bij de meerderheid van de klinisch gediagnosticeerde ATS-patiënten werd een mutatie in KCNJ2 gemeld (63). KCNJ2 codeert een porievormende subeenheid van naar binnen rectificerende kaliumkanalen (Ik1) (figuur 1) (64, 65).
LQTS8
ook bekend als Timothy syndrome (TS), vertonen patiënten ernstige QT-verlenging op hun ECG ‘ s, wat in 100% van de gevallen wordt gecombineerd met syndactylie, kaalheid bij de geboorte en kleine tanden en minder penetrerende cardiale structurele misvormingen, mentale retardatie, autisme en faciale dysmorfe kenmerken (66). Er zijn twee subtypes: TS1 (klassiek) en TS2 (zeldzame vorm).
TS2 is cardiologisch ernstiger dan TS1 (66, 67). TS2-patiënten hebben ook een gebrek aan syndactylie (67). Mutaties in de α-1 subeenheid van de l-type calciumstroom (ICa-L) die codeert voor het gen CACNA1C leiden tot beide vormen van TS (LQTS8). Er zijn alternatief verbonden, wederzijds exclusief exon-8 in het CACNA1C gen, voor duidelijkheid, worden zij genoemd als exon-8 en exon-8A. in hart en hersenen, waar CACNA1C hoofdzakelijk wordt uitgedrukt, wordt exon-8 Gevonden in ∼80% van de mRNA transcripten en exon-8A is aanwezig ∼20% transcripten (66). G406r-mutatie in exon-8A is Causaal voor de klassieke vorm van TS (TS1) en G402S in exon-8 werd gemeld bij severer in TS2. Een nieuwe toevoeging aan deze lijst is de ala1473gly-mutatie, die is beschreven bij een ts-zuigeling met een verder uitgebreid fenotype (68). Alle mutaties waren ofwel de novo of mozaïek in de ouder, en alle resultaat winst van de functie van het ICa-L kanaal (66-69).
LQTS9 en LQTS10
mutaties in CAV3 of SCN4B veroorzaken functieversterking in late INa, wat een lqts3-achtig fenotype (70-74) veroorzaakt. Ze staan bekend als LQTS9 (geassocieerd met CAV3-mutatie) en LQTS10 (geassocieerd met SCN4B-mutatie).
Caveolae worden mooi beschreven door Engelman et al. (72) als “kleine grotten” in het plasmamembraan. Zij zijn kleine ongecoate putten en worden beschouwd als de plaats van belangrijke dynamische en regulerende gebeurtenissen op het plasmamembraan (72, 73). Caveolines zijn de belangrijkste proteã nen in de caveolae, en caveolin-3 (gecodeerd door Gen CAV3) wordt specifiek gevonden in cardiomyocytes en skeletspiercellen. Verschillende cardiale ionenkanalen zijn specifiek gemeld te worden gelokaliseerd in de caveolae uit cardiale myocyten die zijn verrijkt met caveoline-3 (72, 73). Bovendien zijn de componenten van de β-adrenerge receptor signalerende cascade ook aanwezig in met caveolae verrijkte membranen (72, 73).
SCN4B codeert voor NaVß4, een hulp-β-subeenheid van het cardiale natriumkanaal. Tot nu toe is in dit gen slechts één mutatie (L179F) gemeld in een Mexicaanse familie met meerdere aangetaste familieleden (74). Mutatie in dit gen bleek te resulteren in functieversterking van de Nav1.5 stroom (74).
LQTS11
in het hart wordt de sympathische regulatie van de duur van de cardiale actiepotentiaal (APD) gemedieerd door β-adrenerge receptor (β-AR) activering, waarvoor akap9 (Yotiao) moet worden geassembleerd met de α-subeenheid (KvLQT1) van het iks-kanaal. Mutatie in AKAP9 veroorzaakt LQTS11 (75). Tot op heden is slechts één mutatie, S1570L, in AKAP9 gemeld (75).
LQTS12
mutatie in het α-1-Syntrofine (SNTN1) gen is Causaal voor LQTS12. Mutatie in dit gen leidt tot verbetering van de functie van het cardiale natriumkanaal (Nav1.5), wat de pathologische basis is van LQTS12 (76).
LQTS13
g eiwitgekoppeld, inwendig rectificerende kaliumkanaalsubeenheid (Kir3. 4) wordt gecodeerd door het kcnj5-gen. Een functieverlies mutatie in dit gen kan LQTS13 (77) veroorzaken. Tot nu toe is slechts één mutatie, G387R, beschreven in een Chinese familie met negen patiënten met deze mutatie. Verminderde plasma-membraanexpressie van Kir3. 4 werd gesuggereerd als de pathologie van LQTS bij de patiënten.
verworven LQT ‘s
naast de aangeboren LQT’ s bestaat er ook een andere variant van lqt ‘s, bekend als alqt’ s, die wordt veroorzaakt door factoren en stoffen die de kaliumflux verminderen en het vermogen van het myocardium om te repolariseren aantasten. Goed erkende aandoeningen zijn vrouwelijk geslacht, hypokaliëmie en geneesmiddelen die cardiale kaliumkanalen remmen (78-80). Een aantal vaak voorgeschreven geneesmiddelen kan ook bij voorkeur binden en blokkeren het HERG-kanaal (Kv11. 1, een eiwit gecodeerd door kcnh2 gen) en predisponeert voor aLQTS (79, 80). Onlangs werd aangetoond dat blokkade van IK ‘s ook kan bijdragen aan drugs geïnduceerde alqt’ s, vooral wanneer repolarisatiereserve wordt gecompromitteerd (81). Fluoxetine en norfluoxetine bleken de IKS-eigenschappen te onderdrukken, zowel in vivo als in vitro en leidden tot duidelijke LQTS (81). Polymorfismen, D85N in minK (gen KCNE1), T8A, Q9E in MiRP1 (gen KCNE2), die vermeende β-subeenheden van de IKS-en IKr-kanalen zijn, zouden aLQTS veroorzaken (82, 83). Auto-immune LQTS is ook gemeld bij een patiënt met IgG die anti-HERG antilichamen bevat (84).Elektrocardiografische kenmerken in de drie veel voorkomende vormen van lang QT-syndroom
typische ST-T-golfpatronen zijn aanwezig bij de meerderheid van de genotypeerde LQTS-patiënten en kunnen worden gebruikt om lqts1, LQTS2 en mogelijk lqts3-genotypen te identificeren (85, 86).
de lqts1 vorm van de LQTS wordt geassocieerd met een brede T-golf zonder verkorting van het QT-interval tijdens inspanning (figuur 2A). LQTS2 wordt geassocieerd met lage amplitude, vaak bifid, T golven (figuur 2B). LQTS3 wordt geassocieerd met een lang iso-elektrisch segment en een smalle, hoge T-golf (figuur 2C). Pauzeafhankelijkheid van het begin van TdP bij congenitale LQTS is genotype-specifiek, overheersend in LQTS2 maar bijna afwezig in LQTS1 (87).

Figuur 2. Elektrocardiogram opnames van patiënten met LQTS1, LQTS2 en LQTS3. A) 12-lood ECG van een 18-jarige man met een kcnq1-mutatie. Het QT-interval is verlengd (QTc = ±500 ms). Het ST-segment heeft een brede basis en een relatief grote amplitude. Het geleidingsinterval is normaal (standaardkalibratie). B) 12-lead ECG van een 14-jarig meisje met een kcnh2-mutatie. Het QT-interval is verlengd (QTc ± 520 ms). Het ST-segment is gekerfd in lead V3 en heeft een relatief lage amplitude in de extremiteit leads. Het geleidingsinterval is normaal (standaardkalibratie). C) 12-lood ECG van een 12-jarige jongen met een scn5a-mutatie. Het QT-interval is verlengd (QTc ± 600 ms). Het ST-segment heeft een lang (bijna) iso-elektrisch segment met een grote, scherpe en smalle T-golf. Het geleidingsinterval is normaal (standaardkalibratie).
hoewel patronen een specifiek genotype van de LQT ‘ s kunnen suggereren, zijn er vaak uitzonderingen gevonden.
Genotype-fenotype
lang QT-syndroom is een autosomaal dominante ziekte. Genotype-en fenotype analyse onder de heterozygote mutatiedragers is vrij uitgebreid uitgevoerd bij de lqts1, LQTS2 en lqts3 patiënten. Tijdens de kindertijd is het risico op cardiale voorvallen significant hoger bij lqts1-mannen dan bij lqts1-vrouwen, terwijl er geen significante gendergerelateerde verschillen in het risico op cardiale voorvallen werden waargenomen bij lqts2-en lqts3-patiënten (88-91). Tijdens de volwassenheid (ook na de leeftijd van 40 jaar) kunnen lqts1-en lqts2-vrouwen een significant hoger risico op cardiale voorvallen hebben dan de respectievelijke mannen (88-91). Over het algemeen lijkt de letaliteit van cardiale voorvallen overheersend te zijn bij LQTS3-patiënten dan bij lqts1-en LQTS2-patiënten (91). Vrouwen met LQTS hebben een verminderd risico op cardiale gebeurtenissen tijdens de zwangerschap, maar het risico neemt vrij toe tijdens de 9 maanden postpartum periode, vooral bij vrouwen met mutatie in het kcnh2 gen (92).
plotselinge hartdood bij kinderen kan ook worden veroorzaakt door mutaties in de cardiale ionenkanaalgenen (93-96). Ongeveer 28% van de kinderen met een onverklaarbare SCD (tussen 1 en 18 jaar, gemiddelde leeftijd: 12,3 ± 3,8 jaar) waren dragers van mutaties in LQTS causale genen (97). Bij SID ‘ s leken mutaties in SCN5A overheersend (98, 99), maar mutaties in KCNQ1, KCNH2, KCNE2 en CAV3, SCN4B en SCN3B werden ook gevonden (100, 101). Intra-uteriene foetale sterfte werd ook gemeld als gevolg van defecten in cardiale ion channel genen (12, 102).
klinische behandeling van LQTS
stopzetting van alle geneesmiddelen waarvan bekend is dat ze het QT-interval verlengen en ook de correctie van elektrolytenonevenwichtigheden en/of het neerslaan van metabole aandoeningen dienen de primaire focus te zijn tijdens de behandeling van (verworven -) LQTS-patiënten. Symptomen bij LQT’ s worden vaak adrenergisch gemedieerd, daarom wordt over het algemeen een beperking van de deelname van patiënten aan atletische activiteiten Aanbevolen (24, 25). De steunpilaar van de klinische therapie voor de LQTS is β-blokkade. Langwerkende preparaten van propranolol, nadolol en metoprolol worden meestal gebruikt, en hun werkzaamheid in β-blokkade wordt beoordeeld door afzwakken van de hartslag van de oefening (bijv., door >20%) (24, 25). Van alle β-blokkers worden propranolol en nadolol bij symptomatische patiënten als superieur beschouwd dan metoprolol (103). Bovendien kan β-blokkade ook worden gebruikt als profylactische behandeling bij silent mutation dragers om SCD te verminderen (24). In een studie van Barsheshet et al. (30), vertoonden patiënten met C-loop missense-mutaties in het kcnq1-gen een hoog risico op levensbedreigende cardiale gebeurtenissen en hadden zij significante voordelen van de behandeling met β-blokkers. Aangezien vrouwen met LQTS2 een verhoogd risico hebben tijdens de postpartumperiode van 9 maanden, moeten β-blokkers worden voorgeschreven om cardiale voorvallen tijdens deze periode met hoog risico te verminderen (92). Een implanteerbare cardioverter-defibrillator (ICD ‘ s) kan worden overwogen voor patiënten met recidiverende syncope ondanks β-Blokker therapie of bij patiënten met een hoog risico op hartstilstand (bijv. symptomatische lqts2 en LQTS3 met een gedocumenteerde QTc-verlenging). Left cardiac sympathic denervation (LCSD) wordt aanbevolen voor hoog risico LQTS patiënten bij wie een ICD is gecontra-indiceerd of geweigerd en β-blokkers zijn ofwel niet effectief, niet verdragen, niet geaccepteerd of gecontra-indiceerd (18, 104). JLNS-patiënten hebben gewoonlijk een QTc > 500 ms en lopen ook een hoog risico, β-blokkers hebben onvoldoende werkzaamheid bij dergelijke patiënten bij wie een vroege behandeling met ICD werd aanbevolen (18, 31).
LQTS: Saudi Perspective
het eerste rapport over LQT ‘ s uit Saudi-Arabië werd in 1993 gepubliceerd in het Riyadh Armed Forces Hospital (105). Vier zuigelingen en jonge kinderen tussen 6 en 48 maanden met een voorgeschiedenis van recidiverende aanvallen, afkomstig uit één enkele familie, werden gediagnosticeerd als LQTS (105). Familiegeschiedenis toonde aan dat twee-andere uitgebreide familieleden soortgelijke episodes van plotseling verlies van bewustzijn hadden en drie-familieleden stierven plotseling (105). In alle gevallen was de eerste diagnose epilepsie (105). Enkele jaren later werden twee sporadische casusrapporten gerapporteerd met een relatief zwaardere variant van neonatale LQT ‘ s gecombineerd met 2:1 AV-blok (106, 107). Al deze gepubliceerde klinische rapporten waren zonder enige genetische bevindingen die de pathofysiologie van LQTS in hen konden verklaren (105-107).
we hebben voor het eerst genetische defecten gerapporteerd als een pathologische basis van LQT ‘ s bij vergelijkbare patiënten uit Saoedi-Arabië. We hebben zes-Saoedische families onderzocht met een geschiedenis van syncope en plotselinge onverklaarbare sterfgevallen van foetus, pasgeborenen en kinderen (12-14). Autosomaal recessieve LQTS1 werd gediagnosticeerd bij kinderen uit twee families (Figuur 3). Autosomaal recessieve LQTS2 werd gediagnosticeerd in twee families (Figuur 4). In één familie werd bij een vrouwelijke patiënt autosomaal dominante LQTS2 gediagnosticeerd (Figuur 5), de patiënt had syncopale aanvallen tijdens de postpartum herstelperiode in het ziekenhuis, wat zeer gebruikelijk is bij vrouwen die kcnh2-mutatie dragen. In al onze patiënten, identificatie van pathogene mutaties in de LQTS causale cardiale ion kanaal genen leidde tot een bevestigde klinische diagnose, die verkeerd werden gediagnosticeerd als epileptische convulsies voordat ze naar ons worden verwezen (13, 14) onlangs, Shinwari et al. (15) uit het King Faisal Specialist Hospital and Research Centre rapporteerde een lqts1 causale kcnq1 mutatie, H258P, in een grote familie met 12 getroffen individuen. Slechts twee dragers waren symptomatisch, hun QTc was >500 ms en β-blokkers onderdrukten de klinische symptomen bij één patiënt en de tweede symptomatische patiënt had een ICD nodig.
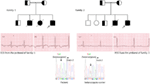
Figuur 3. Stamboom tekening van de familie-1 en 2 met autosomaal recessieve LQTS1, probands worden weergegeven met een pijl. ECG ‘ s van de probands in twee families worden weergegeven in het midden, met een QTc van 557 en 529 ms, respectievelijk. Intronische mutatie, c. 387 -5 T > A in het kcnq1 gen werd gevonden bij de patiënten (getoond in de bodem). Mutatie werd aangetoond met een pijl. Niet-gevulde cirkels en vierkanten zijn geen dragers voor de mutatie. Getroffen individuen worden weergegeven als gevulde Cirkels (vrouwelijk) en vierkanten (Mannelijk). Halfgevulde vierkanten en cirkels zijn individuen met heterozygote mutatie. Overleden individuen worden aangeduid met schuine strepen, probands worden aangeduid met een pijl en bloedverwantschap huwelijk wordt aangegeven door = Exon − intron grens wordt aangegeven door een stippellijn met pijl naar exon.
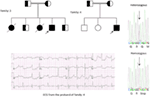
Figuur 4. Stamboom tekening van de familie-3 en 4 met autosomaal recessieve LQTS2. ECG van de proband uit family-4 is weergegeven onder de stamboomtekening, die sinustachycardie toont, bijna volledig AV-blok, breed complex ontsnappingsritme met zeer lange QTc-intervallen (QT >600 ms). Mutatie, c. 3208 C > T (p. Q1070X) in het gen KCNH2 werd gevonden bij de patiënten (rechts afgebeeld), gemarkeerd met een pijl.
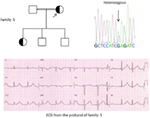
Figuur 5. Linksboven: stamboom van de familie-5. Getroffen individu wordt weergegeven door gevulde cirkel (vrouwelijke) Proband wordt aangegeven door een pijl. 12-lead ECG van de proband (I: 2). ECG toont een hoge piek, een brede basis, een grote amplitude van de T-golf, QTc-interval is 580 ms. Screening van de KCNH2 gen toont vervanging van nucleotide “G” voor Een “a” (c.2362G > A, pijl gemarkeerd), wat leidt tot een aminozuur substitutie, blz.E788K.
Genetische en klinische bevindingen in ons onderzoek (12-14) van Saoedi-Arabië zijn heel intrigerend om verschillende redenen: (1) In totaal hebben we onderzocht zes gezinnen, onder hen vier waren homozygoot/compound heterozygoot voor de mutaties en mutaties die is ontstaan uit een oude bron; (2) alle mutaties in LQT ‘ s waren nieuw, alleen gemeld in deze Arabische families; (3) vanwege de homozygositeit of samengestelde heterozygositeit voor de mutaties, waren klinische fenotypen ook ernstig in onze bestudeerde families (12-14). We stelden voor dat de genetische en fenotypische waarnemingen voortkwamen uit het extreem hoge aantal bloedverwantshuwelijken in Saoedi-Arabië (108, 109). Onze studie leverde het eerste wetenschappelijke bewijs over de rol van bloedverwantschap in het uitoefenen van een centrale rol in onverklaarbare hartritmestoornissen en SCD ‘ s bij kinderen en adolescenten in Saoedi-Arabië (12-14). We hebben ook aangetoond dat de kcnq1 mutatie c.387 -5 T > A (Nm_000218) (Figuur 3) zich in de Assir provincie Saudi-Arabië had verspreid vanuit een gemeenschappelijke voorouder gedurende meerdere generaties als gevolg van hoge incidentie van bloedverwantshuwelijken . Tot nu toe is dit de meest voorkomende lqts1 causale mutatie in Saoedi-Arabische bevolking, die ook werd waargenomen in het King Faisal Specialist Hospital en in Khamis Mashayt Military Hospital (ongepubliceerd). Een vergelijkbaar resultaat werd ook verkregen voor lqts2 causale mutatie, c.3208 C > T (p.Q1070X) (Figuur 4) in het kcnh2 gen (12, 14), dat ook een stichter mutatie is in Saoedi-Arabië en gedurende vele generaties gescheiden is in de Assir regio (zie de kaart, Figuur 6). We voorspellen dat er een aanzienlijk aantal individuen met de genoemde voorouderlijke/stichter mutaties (zowel in KCNQ1 en KCNH2 en andere genen) in deze regio en ook in de grote steden als Riyad, Jeddah en Dammam als gevolg van stedelijke migratie. Meer stichter of voorouderlijke mutaties pathogeen voor LQT ‘ s zijn zeer veel voorzien in andere provincies van Saoedi-Arabië als gevolg van het hoge percentage van bloedverwantschap huwelijken.
Figuur 6. Kaart van Saoedi-Arabië (hoffelijkheid: Wikipedia). Assir regio is gemarkeerd met dikke lijn, waar we de stichter mutaties in KCNQ1 en kcnh2 genen hebben gevonden, beschreven in familie 1-4.
aangezien LQTS een autosomaal dominante ziekte is, werd verwacht dat we voornamelijk heterozygote mutatiedragerpatiënten zouden waarnemen. Maar in onze studie identificeerden we vooral patiënten met recessieve mutaties (12-14). Deze patiënten werden duidelijk eerst symptomatisch en werden voornamelijk doorverwezen naar het hartcentrum van Prins Sultan, waardoor ze onder onze aandacht kwamen. Niettemin, bevindingen uit onze onderzoeken impliceert aan het feit dat recessieve LQTS fatale klinische fenotypen bij kinderen zou kunnen hebben, en ze zijn niet ongewoon in Saoedi-Arabië (12-14). Aangezien de heterozygote mutatiedragers ook gevoelig zijn voor het ontwikkelen van aritmie en de complicaties ervan, moet gezamenlijk initiatief worden genomen om de lokale huisartsen, cardiologen en klinische genetici in een gemeenschappelijk platform te brengen om de personen die risico lopen te identificeren. Verder, op dit moment, hebben we ook geen genetische informatie voor andere familiale aritmie stoornissen, b.v., CPVT, SQTS, BrS, AF, enz. Gespecialiseerde cardiogenetische centra moeten het initiatief nemen om te zoeken naar de genetische defecten, mutaties, en het uitvoeren van genotype-fenotype studies in alle vormen van erfelijke aritmieën. Er moet ook rekening mee worden gehouden dat niet alle genetische aritmieën een familiegeschiedenis zouden hebben, aangezien in veel gevallen de oorzakelijke mutatie van aritmie de novo van oorsprong is, d.w.z. de proband is de eerste patiënt in die familie met de mutatie en hij/zij is de bron om de mutatie in downstream generaties over te brengen (110, 111). Vanwege het zeer hoge aantal verwantschapshuwelijken in Saoedi-Arabië, verwachten we dat veel meer stichtermutaties een cruciale rol spelen bij aangeboren hartritmestoornissen in dit land. Het identificeren van deze stichtermutaties zou onze eerste en belangrijkste taak moeten zijn, die ons zou helpen bij het ontwikkelen van een effectieve pre-symptomatische genetische counseling voor het huwelijk in dit land. Mutaties in LQTS causale genen kunnen variabiliteit in klinische penetrantie veroorzaken, in het ene uiterste kunnen sommige dragers vermoedelijk volledig gezond zijn, maar sommige dragers kunnen hun eerste manifestatie van de ziekte hebben als syncope of plotselinge dood. Plotselinge dood van een jong kind of volwassene stelt een groot deel van psychologische en emotionele last op het gezin, screening van de dragers zijn essentieel aangezien er eenvoudige medicatie, b.v., β-blokkers (ook gedragswijziging) bestaan, die zeer effectief de dragers van de fatale gevolgen van aritmie en SCDs zouden kunnen verhinderen. Personen met homozygote mutaties in de LQTS causale genen kunnen ernstige morbiditeit en extreem hoge sterftecijfers hebben, waaronder foetale brady-tachyaritmieën, en in veel gevallen miskramen, spontane abortussen bij de zwangere moeders (12-14).
er is ook niet veel onderzoek gedaan in Saoedi-Arabië naar de prevalentie van verschillende SNP ‘ s in de aritmiegebonden genen en ook in de genen die hen reguleren. Splawski et al. (112) beschreef een veel voorkomende s1103y variant in het scn5a gen geassocieerd met aritmie in Afro-Amerikanen. Het variant allel genaamd Y1103 is verantwoordelijk voor het versnellen van kanaalactivering, waardoor de kans op hartritmestoornissen bij personen van Afrikaanse afkomst wordt vergroot (52, 112). K393N is een variant in het kcnq1 gen, gerapporteerd bij LQTS1 patiënten in de VS, maar in de Arabische populatie hebben we deze variant ontdekt bij 2% van de individuen (ongepubliceerde gegevens). Of de k393n variant in het kcnq1 gen in Arabieren vergelijkbaar is met de s1103y (SCN5A) variant in Afro-Amerikanen of de d85n (KCNE1) variant in de Japanse bevolking kan ook onderzoek verdienen (59).
belangenverstrengeling verklaring
de auteurs verklaren dat het onderzoek werd uitgevoerd zonder enige commerciële of financiële relatie die als een potentieel belangenconflict kon worden opgevat.
Dankbetuigingen
onze oprechte dank aan Prof. Arthur Wilde, Prof.Connie Bezzina, en de uitgever van het tijdschrift “Heart” voor hun toestemming om de figuur 1 in dit manuscript te gebruiken uit Ref. (113).
1. Bhuiyan ZA. Klinisch en genetisch Spectrum van erfelijke hartritmestoornissen syndromen. Amsterdam: Universiteit Van Amsterdam (2009).
2. Priori SG, Aliot E, Blømstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, et al. Task force on sudden cardiac death, European society of cardiology. Europace (2002) 4: 3-18. doi: 10.1053 / eupc.2001.0214
CrossRef Volledige Tekst
3. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, et al. Scn5a mutaties geassocieerd met een erfelijke hartritmestoornis, lang QT-syndroom. Cell (1995) 80: 805-11. doi:10.1016/0092-8674(95)90359-3
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
4. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. Een moleculaire basis voor hartritmestoornissen: Herg-mutaties veroorzaken lang QT-syndroom. Cell (1995) 80: 795-803. doi: 10.1016/0092-8674(95)90358-5
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
5. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, vanraay TJ, et al. Positioneel klonen van een nieuw kaliumkanaalgen: kvlqt1-mutaties veroorzaken hartritmestoornissen. Nat Genet (1996) 12: 17-23. doi: 10.1038 / ng0196-17
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
6. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutaties in het hmink-gen veroorzaken het lange QT-syndroom en onderdrukken de IKs-functie. Nat Genet (1997) 17: 338-40. doi: 10.1038 / ng1197-338
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
7. Sanguinetti MC, Jiang C, Curran ME, Keating MT. Een mechanistische link tussen een erfelijke en een verworven hartritmestoornis: HERG codeert het IKr kaliumkanaal. Cell (1995) 81:299-307. doi:10.1016/0092-8674(95)90340-2
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. Een nieuwe mutatie in het kaliumkanaalgen KVLQT1 veroorzaakt het cardioauditory syndroom van Jervell en Lange-Nielsen. Nat Genet (1997) 15: 186-9. doi: 10.1038 / ng0297-186
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
9. Kucera JP, Rohr S, Rudy Y. lokalisatie van natriumkanalen in intercalated disks moduleert hartgeleiding. Circ Res (2002) 91: 1176-82. doi: 10.1161 / 01.RES. 0000046237. 54156. 0 A
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
10. Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Connexin43 remodellering veroorzaakt door remming van plakophilin – 2 expressie in cardiale cellen. Circ Res (2007) 101: 703-11. doi: 10.1161 / CIRCRESAHA.107.154252
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
11. Rohr S. moleculaire kruisverwijzing tussen mechanische en elektrische kruispunten op de tussenliggende schijf. Circ Res (2007) 101: 637-9. doi: 10.1161 / CIRCRESAHA.107.161901
CrossRef Volledige Tekst
12. Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, et al. Recidiverend intra-uteriene foetaal verlies als gevolg van bijna afwezigheid van HERG: klinische en functionele karakterisering van een homozygote nonsense HERG Q1070X mutatie. Heart Rhythm (2008) 5: 553-61. doi: 10.1016/j.hrthm.2008.01.020
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
13. Bhuiyan ZA, Momenah TS, Amin TS, Al-Khadra AS, Alders M, Wilde AAM, et al. Een Intronische mutatie die leidt tot onvolledige overslaan van exon-2 in KCNQ1 redt het gehoor bij het syndroom van Jervell en Lange-Nielsen. Prog Biophys Mol Biol (2008) 98: 319-27. doi: 10.1016 / j. pbiomolbio.2008.10.004
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
14. Bhuiyan ZA, Al-Shahrani s, Al-Khadra AS, Al-Ghamdi s, Al-Kalaf K, Mannens MMAM, et al. Klinische en genetische analyse van het lange QT-syndroom bij kinderen uit zes families in Saoedi-Arabië: zijn ze verschillend? Pediatr Cardiol (2009) 30: 490-501. doi: 10.1007 / s00246-008-9377-y
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
15. Shinwari ZM, Al-Hazzani A, Dzimiri N, Tulbah S, Mallawi Y, al-Fayyadh M, et al. Identificatie van een nieuwe kcnq1-mutatie in een grote Saoedische familie met lang QT-syndroom: klinische gevolgen en preventieve implicaties. Clin Genet (2013) 83: 370-4. doi: 10.1111 / j. 1399-0004. 2012. 01914.X
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalentie van het congenitale lange-QT-syndroom. Circulation (2009) 120:1761-7. doi: 10.1161 / CIRCULATIONAHA.109.863209
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
17. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostische criteria voor het lange QT-syndroom. Update. Circulation (1993) 88: 782-4. doi: 10.1161 / 01.CIR.88.2.782
CrossRef Volledige Tekst
18. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. Samenvatting: hrs/EHRA / APHRS consensusverklaring over de diagnose en behandeling van patiënten met erfelijke primaire aritmiesyndromen. Europace (2013) 15: 1389-406. doi: 10.1093 / europace / eut272
CrossRef volledige tekst
19. Liu JF, Jons C, Moss AJ, McNitt S, Peterson DR, Qi M, et al. Syndrome Register. Risicofactoren voor recidiverende syncope en daaropvolgende fatale of bijna fatale voorvallen bij kinderen en adolescenten met een lang QT-syndroom. J Am Coll Cardiol (2011) 57: 941-50. doi: 10.1016 / j.jacc.2010.10.025
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
20. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spectrum van mutaties in genen voor het lange-QT-syndroom. KVLQT1, HERG, SCN5A, KCNE1 en KCNE2. Oplage (2000) 102:1178-85. doi: 10.1161 / 01.CIR.102.10.1178
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
21. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Samengestelde mutaties: een veel voorkomende oorzaak van ernstig QT-syndroom. Oplage (2004) 109:1834-41. doi: 10.1161 / 01.CIR.0000125524.34234.13
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
22. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, et al. Risico op levensbedreigende cardiale voorvallen bij patiënten met lang QT-syndroom en meerdere mutaties. Hartritme (2013) 10: 378-82. doi: 10.1016/j.hrthm.2012.11.006
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
23. Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Genetische tests in het lange QT-syndroom: ontwikkeling en validatie van een efficiënte benadering van genotypering in de klinische praktijk. JAMA (2005) 294: 2975-80. doi: 10.1001 / jama.294.23.2975
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
24. Roden DM. Klinische praktijk. Long-QT syndroom. N Engl J Med (2008) 358:169-76. doi: 10.1056 / NEJMcp0706513
CrossRef volledige tekst
25. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-fenotype correlatie bij het lange-QT-syndroom: genspecifieke triggers voor levensbedreigende aritmieën. Circulation (2001) 103:89-95. doi: 10.1161 / 01.CIR.103.1.89
CrossRef Volledige Tekst
26. Ackerman MJ, tester DJ, Porter CJ. Zwemmen, een gen-specifieke aritmogene trigger voor erfelijk lang QT syndroom. Mayo Clin Proc (1999) 74: 1088-94. doi:10.4065/74.11.1088
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
27. Kapa s, tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, et al. Genetische tests voor het lange-QT-syndroom: het onderscheiden van pathogene mutaties van goedaardige varianten. Circulation (2009) 120: 1752-60. doi: 10.1161 / CIRCULATIONAHA.109.863076
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
28. Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, et al. Klinische aspecten van type 1-lang QT-syndroom naar locatie, codeertype en Biofysische functie van mutaties waarbij het kcnq1-gen betrokken is. Circulation (2007) 115:2481-9. doi: 10.1161 / CIRCULATIONAHA.106.665406
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
29. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. Varianten in het 3 ‘ onvertaalde gebied van het kcnq1-gecodeerde KV7.1 kaliumkanaal wijzigen de ernst van de ziekte bij patiënten met type 1 lang QT-syndroom op een allel-specifieke manier. Eur Heart J (2012) 33: 714-23. doi: 10.1093 / eurheartj / ehr473
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
30. Barsheshet A, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu W, et al. Mutaties in cytoplasmatische lussen van het KCNQ1-kanaal en het risico op levensbedreigende voorvallen: implicaties voor mutatie-specifieke respons op β-Blokker therapie bij type 1 lang QT syndroom. Oplage (2012) 125: 1988-96. doi: 10.1161 / CIRCULATIONAHA.111,048041
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
31. Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. Het syndroom van Jervell en lange-Nielsen: natuurlijke geschiedenis, moleculaire basis en klinische uitkomst. Circulation (2006) 113:783-90. doi: 10.1161 / CIRCULATIONAHA.105.592899
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
32. Bhuiyan ZA, Wilde AA. IKs in hart en gehoor, het oor kan doen met minder dan het hart. Circ Cardiovasc Genet (2013) 6:141-3. doi: 10.1161 / CIRCGENETICS.113.000143
CrossRef Volledige Tekst
33. Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Pathofysiologische mechanismen van dominante en recessieve kvlqt1 K+ – kanaalmutaties gevonden in erfelijke hartritmestoornissen. Hum Mol Genet (1997) 6: 1943-9. doi: 10.1093 / hmg / 6.11.1943
CrossRef Volledige Tekst
34. Wilde AA, Jongbloed RJ, Doevendans PA, Düren DR, Hauer RN, van Langen IM, et al. Auditieve stimuli als trigger voor aritmische gebeurtenissen onderscheiden HERG-gerelateerde (LQTS2) patiënten van kvlqt1-gerelateerde patiënten (LQTS1). J Am Coll Cardiol (1999) 33: 327-32. doi: 10.1016/S0735-1097(98)00578-6
volledige tekst CrossRef
35. Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, et al. Verhoogd risico op aritmische voorvallen bij het lange-QT-syndroom met mutaties in het poriegebied van het humane ether-a-go-go-gerelateerd genkaliumkanaal. Circulation (2002) 105:794-9. doi: 10.1161 / hc0702. 105124
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
36. Lupoglazoff JM, Denjoy I, Villain E, Fressart V, Simon F, Bozio A, et al. Lang QT-syndroom bij pasgeborenen: geleidingsstoornissen geassocieerd met HERG-mutaties en sinusbradycardie met kcnq1-mutaties. J Am Coll Cardiol (2004) 43: 826-30. doi: 10.1016 / j.jacc.2003.09.049
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
37. Chevalier P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ, et al. Torsades de pointes complicerende atrioventriculair blok: bewijs voor een genetische aanleg. Heart Rhythm (2007) 4: 170-4. doi: 10.1016/j.hrthm.2006.10.004
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
38. Hoorntje T, Alders M, van Tintelen P, Van der Lip K, Sreeram N, van der Wal A, et al. Homozygote voortijdige afkaping van het Herg-eiwit: de menselijke Herg knockout. Circulation (1999) 100: 1264-7. doi: 10.1161 / 01.CIR.100.12.1264
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
39. Piippo K, Laitinen P, Swan H, Toivonen L, Viitasalo M, Pasternack M, et al. Homozygositeit voor een Herg kalium kanaal mutatie veroorzaakt een ernstige vorm van lange QT-syndroom: identificatie van een schijnbare stichter mutatie in de Finnen. J Am Coll Cardiol (2000) 35: 1919-25. doi: 10.1016/S0735-1097(00)00636-7
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
40. Johnson WH Jr, Yang P, Yang T, Lau YR, Mostella BA, Wolff DJ, et al. Klinische, genetische en Biofysische karakterisering van een homozygote Herg-mutatie die ernstig neonataal lang QT-syndroom veroorzaakt. Pediatr Res (2003) 53:744-8. doi: 10.1203 / 01.PDR.0000059750.17002.B6
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
41. Ackerman MJ, tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Etnische verschillen in cardiale kaliumkanaalvarianten: implicaties voor genetische gevoeligheid voor plotselinge hartdood en genetische tests voor congenitaal lang QT-syndroom. Mayo Clin Proc (2003) 78: 1479-87. doi:10.4065/78.12.1479
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
42. Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T is een genetische modifier van latent congenitaal lang-QT syndroom. Circulation (2005) 112:1251-8. doi: 10.1161 / CIRCULATIONAHA.105.549071
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
43. Nof E, Cordeiro JM, Pérez GJ, Scornik FS, Calloe K, Love B, et al. Een gemeenschappelijk enkelnucleotidepolymorfisme kan het lange-QT type 2-syndroom verergeren, wat leidt tot wiegendood. Circ Cardiovasc Genet (2010) 3: 199-206. doi: 10.1161 / CIRCGENETICS.109.898569
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
44. Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, et al. Een enkele na (+) kanaalmutatie die zowel long-QT als Brugada syndromen veroorzaakt. Circ Res (1999) 85: 1206-13. doi: 10.1161 / 01.RES.85.12.1206
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
45. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, et al. Nieuwe scn5a-mutatie die leidt tot een geïsoleerd hartgeleidingsdefect of het Brugada-syndroom in een grote Franse familie. Circulation (2001) 104:3081-6. doi: 10.1161 / hc5001. 100834
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
46. Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, et al. De e1784k-mutatie in SCN5A is geassocieerd met een gemengd klinisch fenotype van het type 3 lange QT-syndroom. J Clin Invest (2008) 118:2219-29. doi: 10.1172 / JCI34057
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
47. Keller DI, Acharfi S, Delacrétaz E, Benammar N, Rotter M, Pfammatter JP, et al. Een nieuwe mutatie in SCN5A, delQKP 1507-1509, veroorzakend lang QT-syndroom: rol van Q1507 residu in natriumkanaalinactivatie. J Mol Cell Cardiol (2003) 35: 1513-21. doi: 10.1016 / j. yjmcc.2003.08.007
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
48. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risicostratificatie bij het lange-QT-syndroom. N Engl J Med (2003) 348:1866-74. doi: 10.1056 / NEJMoa022147
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
49. Lupoglazoff JM, Cheav T, Baroudi G, Berthet M, Denjoy I, Cauchemez B, et al. Homozygote SCN5A-mutatie bij lang QT-syndroom met functioneel twee-op-één atrioventriculair blok. Circ Res (2001) 89: E16-21. doi: 10.1161 / hh1401. 095087
CrossRef volledige tekst
50. Shi R, Zhang Y, Yang C, Huang C, Zhou X, Qiang H, et al. De cardiale natriumkanaalmutatie delQKP 1507-1509 wordt geassocieerd met het zich uitbreidende fenotypische spectrum van LQT3, geleidingsstoornis, gedilateerde cardiomyopathie en hoge incidentie van plotselinge jeugddood. Europace (2008) 10: 1329-35. doi: 10.1093 / europace / eun202
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
51. Zumhagen s, Veldkamp MW, Stallmeyer B, Baartscheer A, Eckardt L, Paul M, et al. Een heterozygote deletiemutatie in het cardiale natriumkanaalgen SCN5A met verlies – en gain-van-functiekenmerken manifesteert zich als geïsoleerde geleidingsziekte, zonder tekenen van Brugada of lang QT-syndroom. PLoS One (2013) 8: e67963. doi: 10.1371 / journal.pone.0067963
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
52. Plant LD, Bowers PN, Liu Q, Morgan T, Zhang T, State MW, et al. Een gemeenschappelijke cardiale natriumkanaalvariant geassocieerd met wiegendood in Afro-Amerikanen, SCN5A S1103Y. J Clin Invest (2006) 116:430-5. doi: 10.1172 / JCI25618
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
53. Mohler PJ, Schott JJ, Gramolini EA, Dilly KW, Guatimosim S, duBell WH, et al. Ankyrine – B-mutatie veroorzaakt type 4 Long-QT cardiale aritmie en plotselinge hartdood. Nature (2003) 421:634-9. doi: 10.1038 / nature01335
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
54. Schott JJ, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB, et al. Het in kaart brengen van een gen voor het lange QT-syndroom naar chromosoom 4q25-27. Am J Hum Genet (1995) 57: 1114-22.
Pubmed Abstract / Pubmed Volledige Tekst
55. Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and LSK (minK) proteins associate to form the i(Ks) cardiale potassium current. Nature (1996) 384: 78-80. doi: 10.1038 / 384078a0
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
56. Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Coassemblage van K (V)lqt1 en nertseiwitten(IsK) om cardiaal i (Ks) kaliumkanaal te vormen. Nature (1996) 384:80-3. doi: 10.1038 / 384080a0
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
57. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al. Mutaties van KCNE1 veroorzaken het syndroom van Jervell en het syndroom van lange-Nielsen. Nat Genet (1997) 17: 267-8. doi: 10.1038 / ng1197-267
CrossRef volledige tekst
58. Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutatie van het gen voor IKs geassocieerd met zowel jervell en Lange-Nielsen en Romano-Ward vormen van lange-QT-syndroom. Circulation (1998) 97:142-6. doi: 10.1186/1471-2350-9-24
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85N, een kcne1 polymorfisme, is een ziekte-veroorzakende genvariant in het lange QT-syndroom. J Am Coll Cardiol (2009) 54: 812-9. doi: 10.1016 / j.jacc.2009.06.005
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
60. Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, et al. Genetische variaties van KCNQ1, KCNH2, SCN5A, KCNE1 en KCNE2 bij patiënten met een geneesmiddelgeïnduceerd lang QT-syndroom. Mol Med (2004) 82:182-8. doi: 10.1007/s00109-003-0522-Z
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
61. Abbott GW, Sesti F, Splawski I, Buck me, Lehmann MH, Timothy KW, et al. MiRP1 vormt IKr kaliumkanalen met HERG en wordt geassocieerd met hartritmestoornissen. Cell (1999) 97: 175-87. doi: 10.1016 / S0092-8674 (00)80728-X
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
62. Gordon E, Panaghie G, Deng L, Bee KJ, Roepke TK, Krogh-Madsen T, et al. Een kcne2-mutatie bij een patiënt met hartritmestoornissen geïnduceerd door gehoorstimuli en een verstoorde elektrolytenbalans in het serum. Cardiovasc Res (2008) 77: 98-106. doi: 10.1093/cvr / cvm030
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
63. Gips NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, et al. Mutaties in Kir2.1 veroorzaken de ontwikkelings-en episodische elektrische fenotypen van het syndroom van Andersen. Cell (2001) 105:511-9. doi: 10.1016 / S0092-8674(01)00342-7
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primaire structuur en functionele expressie van een muis naar binnen gelijkrichter kalium kanaal. Nature (1993) 362: 127-33. doi: 10.1038 / 362127a0
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
65. Raab-Graham KF, Radeke CM, Vandenberg CA. Moleculair klonen en expressie van een menselijk hart naar binnen gelijkrichter kalium kanaal. Neuroreport (1994) 5: 2501-5. doi: 10.1097/00001756-199412000-00024
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
66. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca (V)1.2 de dysfunctie van het calciumkanaal veroorzaakt een multisystemwanorde met inbegrip van aritmie en autisme. Cell (2004) 119:19-31. doi: 10.1016 / j.cel.2004.09.011
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
67. Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Ernstige aritmiestoornis veroorzaakt door cardiale L-type calciumkanaalmutaties. Proc Natl Acad Sci U S A (2005) 102:8089-96. doi: 10.1073 / pnas.0502506102
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
68. Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L, et al. Lange QT, syndactylie, gewrichtscontracturen, beroerte en nieuwe CACNA1C-mutatie: uitbreiding van het spectrum van Timothy-syndroom. Am J Med Genet A (2012) 158A:182-7. doi: 10.1002/ajmg.a.34355
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
69. Dufendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. Maternaal mosaïcisme verwart de neonatale diagnose van type 1 Timoteüsyndroom. Kindergeneeskunde (2013) 131:e1991–5. doi: 10.1542 / peds.2012-2941
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
70. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. Mutant caveolin-3 induceert aanhoudende late natriumstroom en wordt geassocieerd met het lange-QT-syndroom. Oplage (2006) 114:2104–12. doi: 10.1161 / CIRCULATIONAHA.106.635268
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
71. Cronk LB, Ye B, Kaku T, tester DJ, Vatta M, Makielski JC, et al. Nieuw mechanisme voor wiegendoodsyndroom: aanhoudende late natriumstroom secundair aan mutaties in caveolin-3. Hartritme (2007) 4: 161-6. doi: 10.1016/j.hrthm.2006.11.030
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
72. Engelman JA, Zhang X, Galbiati F, Volonte D, Sottia F, Pestell RG, et al. Moleculaire Genetica van de caveolin genfamilie: implicaties voor menselijke kanker, diabetes, de ziekte van Alzheimer, en spierdystrofie. Am J Hum Genet (1998) 63: 1578-87. doi:10.1086/302172
volledige tekst CrossRef
73. Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Lokalisatie van cardiale L-type CA(2+) kanalen naar een caveolair macromoleculair signalerend complex is vereist voor bèta (2)-adrenerge Regulatie. Proc Natl Acad Sci U S A (2006) 103:7500-5. doi: 10.1073 / pnas.0503465103
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
74. Medeiros-Domingo A, Kaku T, tester DJ, Iturralde-Torres P, Itty A, Ye B, et al. SCN4B-gecodeerde natriumkanaalbeta4 subeenheid bij congenitaal lang QT-syndroom. Circulation (2007) 116:134-42. doi: 10.1161 / CIRCULATIONAHA.106.659086
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
75. Chen L, Marquardt ML, tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutatie van een A-kinase-verankerend eiwit veroorzaakt lang-QT-syndroom. Proc Natl Acad Sci U S A (2007) 104:20990-5. doi: 10.1073 / pnas.0710527105
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
76. Wu G, Ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, et al. Alfa-1-syntrophine mutatie en het lange QT-syndroom: een ziekte van verstoring van het natriumkanaal. Circ Arrhythm Electrofysiol (2008) 1: 193-201. doi: 10.1161/CIRCEP.108,769224
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, et al. Identificatie van een Kir3.4 mutatie bij congenitaal lang QT-syndroom. Am J Hum Genet (2010) 86: 872-80. doi: 10.1016 / j. ajhg.2010.04.017
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
78. Roden DM. Mechanismen en behandeling van proaritmie. Am J Cardiol (1998) 82:49I–57I. doi: 10.1016/S0002-9149 (98)00472-X
CrossRef volledige tekst
79. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. Een structurele basis voor geneesmiddel-geïnduceerd lang QT-syndroom. Proc Natl Acad Sci U S A (2000) 97: 12329-33. doi: 10.1073 / pnas.210244497
CrossRef Volledige Tekst
80. Kannankeril PJ, Roden DM. Drug-induced long QT en torsade de pointes: recente vooruitgang. Curr Opin Cardiol (2007) 22: 39-43. doi: 10.1097 / HCO.0b013e32801129eb
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
81. Veerman CC, Verkerk EA, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC, et al. Langzame vertraagde gelijkrichter kalium huidige blokkade draagt belangrijk bij aan geneesmiddel-geïnduceerde lange QT-syndroom. Circ Arrhythm Electrofysiol (2013) 6: 1002-9. doi: 10.1161/CIRCEP.113.000239
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
82. Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. Een veelvoorkomend polymorfisme geassocieerd met antibioticageïnduceerde hartritmestoornissen. Proc Natl Acad Sci U S A (2000) 97: 10613-8. doi: 10.1073 / pnas.180223197
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
83. Kääb S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, et al. Een groot kandidaat gen onderzoek identificeert het kcne1 d85n polymorfisme als een mogelijke modulator van drug-geïnduceerde torsades de pointes. Circ Cardiovasc Genet (2012) 5: 91-9. doi: 10.1161 / CIRCGENETICS.111.960930
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
84. Nakamura K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase S, et al. Anti-KCNH2 antilichaam-geïnduceerd lang QT-syndroom: nieuwe verworven vorm van lang QT-syndroom. J Am Coll Cardiol (2007) 50: 1808-9. doi: 10.1016 / j.jacc.2007.07.037
CrossRef Volledige Tekst
85. Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, et al. ECG T-golfpatronen in genetisch verschillende vormen van het erfelijke lange QT-syndroom. Oplage (1995) 92:2929-34. doi: 10.1161 / 01.CIR.92.10.2929
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
86. Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, et al. Spectrum van ST-T-golfpatronen en repolarisatieparameters bij congenitaal lang QT-syndroom: ECG-bevindingen identificeren genotypes. Circulation (2000) 102:2849-55. doi: 10.1161 / 01.CIR.102.23.2849
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
87. Tan HL, Bardai a, Shimizu W, Moss AJ, Schulze-Bahr E, Noda T, et al. Genotype-specifiek begin van aritmieën bij congenitaal verlengd QT-syndroom: mogelijke implicaties voor de therapie. Circulation (2006) 114:2096-103. doi: 10.1161 / CIRCULATIONAHA.106.642694
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
88. Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Leeftijd-en geslachtsgerelateerde verschillen in klinische manifestaties bij patiënten met congenitaal lang QT-syndroom: bevindingen uit het internationale LQTS-register. Circulation (1998) 97:2237-44. doi: 10.1161 / 01.CIR.97.22.2237
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
89. Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, et al. Invloed van genotype op het klinische verloop van het lange-QT-syndroom. International Long-QT Syndrome Registry Research Group. N Engl J Med (1998) 339:960-5. doi: 10.1056 / NEJM199810013391404
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
90. Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson DR, McNitt s, et al. Lang QT-syndroom na de leeftijd van 40. Circulation (2008) 117:2192-201. doi: 10.1161 / CIRCULATIONAHA.107.729368
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
91. Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, et al. Syndrome Register. Modulerende effecten van leeftijd en geslacht op het klinische verloop van het lange QT-syndroom naar genotype. J Am Coll Cardiol (2003) 42: 103-9. doi: 10.1016/S0735-1097(03)00554-0
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
92. Seth R, Moss AJ, McNitt S, Zareba W, Andrews ML, Qi M, et al. Lang QT-syndroom en zwangerschap. J Am Coll Cardiol (2007) 49: 1092-8. doi: 10.1016 / j.jacc.2006.09.054
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
93. Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, et al. Een moleculair verband tussen het wiegendoodsyndroom en het lange-QT-syndroom. N Engl J Med (2000) 343:262-7. doi: 10.1056 / NEJM200007273430405
CrossRef volledige tekst
94. Schwartz PJ, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, et al. Moleculaire diagnose bij een kind met wiegendoodsyndroom. Lancet (2001) 358:1342-3. doi: 10.1016 / S0140-6736(01)06450-9
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
95. Christiansen M, Tønder N, Larsen LA, Andersen PS, Simonsen H, Oyen N, et al. Mutaties in het HERG K + – ion kanaal: een nieuw verband tussen lang QT-syndroom en wiegendood syndroom. Am J Cardiol (2005) 95: 433-4. doi: 10.1016 / j. amjcard.2004.09.054
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
96. Tester DJ, Ackerman MJ. Postmortem lange QT syndroom genetische testen voor plotselinge onverklaarbare dood in de jongeren. J Am Coll Cardiol (2007) 49: 240-6. doi: 10.1016 / j.jacc.2006.10.010
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
97. Hofman N, Tan HL, Clur SA, Alders M, Van Langen IM, Wilde AA. Bijdrage van erfelijke hartziekte aan plotselinge hartdood in de kindertijd. Kindergeneeskunde (2007) 120: e967-73. doi: 10.1542 / peds.2006-3751
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
98. Wedekind H, Smits JP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, et al. De novo mutatie in het scn5a gen geassocieerd met een vroeg begin van wiegendood. Circulation (2001) 104:1158-64. doi: 10.1161 / hc3501. 095361
CrossRef volledige tekst
99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, et al. Cardiale natriumkanaaldisfunctie bij wiegendoodsyndroom. Circulation (2007) 115:368-76. doi: 10.1161 / CIRCULATIONAHA.106.646513
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
100. Van Norstrand DW, Valdivia CR, tester DJ, Ueda K, London B, Makielski JC, et al. Moleculaire en functionele karakterisatie van nieuwe glycerol-3-fosfaat dehydrogenase 1-achtige gen (GPD1-L) mutaties in wiegendood syndroom. Circulation (2007) 116:2253-9. doi: 10.1161 / CIRCULATIONAHA.107.704627
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
101. Tan BH, Pundi KN, van Norstrand DW, Valdivia CR, tester DJ, Medeiros-Domingo A, et al. Mutaties in de bèta-subeenheden van het natriumkanaal. Hartritme (2010) 7: 771-8. doi: 10.1016/j.hrthm.2010.01.032
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
102. Miller te, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, et al. Recidiverend foetaal verlies in het derde trimester en maternaal mosaïcisme voor het lange-QT-syndroom. Oplage (2004) 109:3029-34. doi: 10.1161 / 01.CIR.0000130666.81539.9 E
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
103. Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Niet alle bètablokkers zijn gelijk bij de behandeling van lang QT-syndroom type 1 en 2: Hogere recidieven van voorvallen onder Metoprolol. J Am Coll Cardiol (2012) 60: 2092-6. doi: 10.1016 / j.jacc.2012.07.046
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
104. Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Left cardiac sympathic denervation bij de behandeling van hoog-risico patiënten met het lange-QT-syndroom. Oplage (2004) 109:1826-33. doi: 10.1161 / 01.CIR.0000125523.14403.1 E
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
105. Singh B, al Shahwan SA, Habbab MA, al Deeb SM, Biary N. idiopathische lange QT-syndroom: het stellen van de juiste vraag. Het is niet zo moeilijk.:741. doi:10.1016/0140-6736(93)90501-7
volledige tekst CrossRef
106. Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. The long QT syndrome with impaired atrioventricular conduction: a maligne variant in infants. J Cardiovasc Electrophysiol (1998) 9: 1225-32. doi: 10.1111 / j. 1540-8167.1998.tb00096.X
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
107. Kantoch MJ, Qurashi MM, Bulbul ZR, Gorgels AP. Een pasgeborene met een complexe aangeboren hartziekte, atrioventriculair blok en torsade de pointes ventriculaire tachycardie. Pacing Clin Electrophysiol (1998) 21:2664-7. doi: 10.1111 / j. 1540-8159.1998.tb00043.x
CrossRef volledige tekst
108. El-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Bloedverwantschap onder de Saudi-Arabische bevolking. J Med Genet (1995) 32: 623-6. doi: 10.1136 / jmg.32.8.623
CrossRef Volledige Tekst
109. El Mouzan MI, Al Salloum AA, al Herbish AS, Qurachi MM, Al Omar AA. Bloedverwantschap en belangrijke genetische stoornissen bij Saoedische kinderen: een op gemeenschap gebaseerde cross-sectionele studie. Ann Saudi Med (2008) 28: 169-73. doi:10.4103/0256-4947.51726
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
110. Medeiros-Domingo A, Bhuiyan ZA, tester DJ, Hofman N, Bikker H, van Tintelen JP, et al. Het RYR2-gecodeerde ryanodine receptor / calcium release kanaal bij patiënten die eerder gediagnosticeerd zijn met ofwel catecholaminerge polymorfe ventriculaire tachycardie of genotype-negatief, inspanningsgeïnduceerd lang QT-syndroom: een uitgebreide open reading frame mutationele analyse. J Am Coll Cardiol (2009) 54: 2065-74. doi: 10.1016 / j.jacc.2009.08.022
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
111. Al-Aama JY, al-Ghamdi S, Bdier AY, Wilde AA, Bhuiyan ZA. De novo mutatie in het kcnq1-gen veroorzaakt door het syndroom van Jervell en Lange-Nielsen. Clin Genet (2013). doi: 10.1111 / cge.12300
Pubmed Abstract / Pubmed Full Text / CrossRef Full Text
112. Splawski I, Timothy KW, Tateyama M, Clancy CE, Malhotra A, Beggs AH, et al. Variant van scn5a natriumkanaal betrokken bij het risico van hartritmestoornissen. Wetenschap (2002) 297:1333-6. doi: 10.1126 / wetenschap.1073569
CrossRef Volledige Tekst
113. Wilde AA, Bezzina CR. Genetica van hartritmestoornissen. Heart (2005) 91: 1352-8.