mucopolysaccharidoses: early diagnostic signs in infants and children
mucopolysaccharidoses (MPS) zijn een groep van klinisch heterogene ziekten veroorzaakt door deficiënties van de lysosomale enzymen die nodig zijn voor de afbraak van glycosaminoglycanen (GAGs).MPS worden gekenmerkt door progressieve en systemische klinische manifestaties. Ondanks hun biochemische en genetische heterogeniteit delen verschillende typen belangrijke klinische kenmerken in verschillende combinaties, waaronder gewrichts-en skeletdysplasie met stijfheid (behalve MPS IV waar sprake is van laxiteit) en pijn, Grove gelaatstrekken, vertroebeling van het hoornvlies, inguinale of abdominale hernia ‘ s, terugkerende bovenste luchtweginfecties, hartklepziekte, carpaal tunnelsyndroom en variabele neurologische betrokkenheid. Deze kenmerken verschijnen meestal in de eerste levensmaanden voor de ernstige vormen en in de vroege kindertijd voor de meer verzwakte vormen, maar worden vaak onderschat en in het algemeen alleen in aanmerking genomen wanneer duidelijk duidelijk.
de zeldzaamheid van deze aandoeningen en de variabiliteit in de klinische presentatie leiden vaak tot een diagnostische vertraging; dit kan variëren van maanden, voor de ernstige vormen, tot jaren, voor de verzwakte vormen (zie Rigoldi et al. in dit supplement). Niettemin, wegens de snelle vooruitgang en de dringende behoefte aan interventie in de strenge presentaties, kunnen zelfs maanden van vertraging in diagnose catastrofale resultaten voor de toekomstige gezondheid van de patiënt veroorzaken.Het doel is in dit overzicht de zeer vroege tekenen van de ernstige vormen te onderstrepen die de kinderarts zouden moeten waarschuwen bij hun eerste verschijning.
- welke tekenen / symptomen kunnen we verwachten bij kinderen met ernstige vormen van MPS?
- Wat zijn de verschillende presentaties van de verschillende typen parlementsleden?
- MPS met somatische en cognitieve betrokkenheid
- MPS met voornamelijk neurologische en cognitieve betrokkenheid
- MPS met prevalente somatische betrokkenheid
welke tekenen / symptomen kunnen we verwachten bij kinderen met ernstige vormen van MPS?
symptomen en tekenen die wijzen op verschillende vormen van MPS worden gerapporteerd in Tabel 1 naar leeftijd. In de eerste 6 levensmaanden kunnen inguinale hernia ‘ s, abnormaal frequente luchtweginfecties, otitis en organomegalie worden waargenomen bij MPS-patiënten, met name bij MPS I (Hurlersyndroom), dat het meest vroegrijpe ernstige begin heeft. Bovendien ontwikkelen deze zuigelingen van 6 tot 12 maanden heel vaak een gibbus( toraco – lumbale kyfose), hartgeruis, navelbreuk, milde hypotonie en groeivertraging . Ze kunnen ook de typische Grove gezichtsuitdrukking hebben (Fig. 1). MPS II, VI en VII kunnen een soortgelijk vroeg fenotype vertonen. MPS IV-patiënten hebben een vroeg skeletfenotype met het optreden van heupdysplasie in de eerste levensmaanden en duivenkast (pectus carinatum) in de eerste 12-18 maanden. MPS II-zuigelingen vertonen een vergelijkbare betrokkenheid, maar met een latere aanvang. In het tweede levensjaar ontwikkelen MPS I hurlersyndroom-zuigelingen cognitieve vertraging, terwijl een MPS II-patiënt alleen kan worden geïdentificeerd door overgroei, frequente luchtweginfecties en meerdere operaties ; de typische gelaatstrekken verschijnen pas later. Sommige MPS II-patiënten ontwikkelen ook hyperactiviteit tussen 18 en 24 maanden. In het derde levensjaar zijn hyperactiviteit en cognitieve vertraging gemakkelijk herkenbaar in MPS II en MPS III.
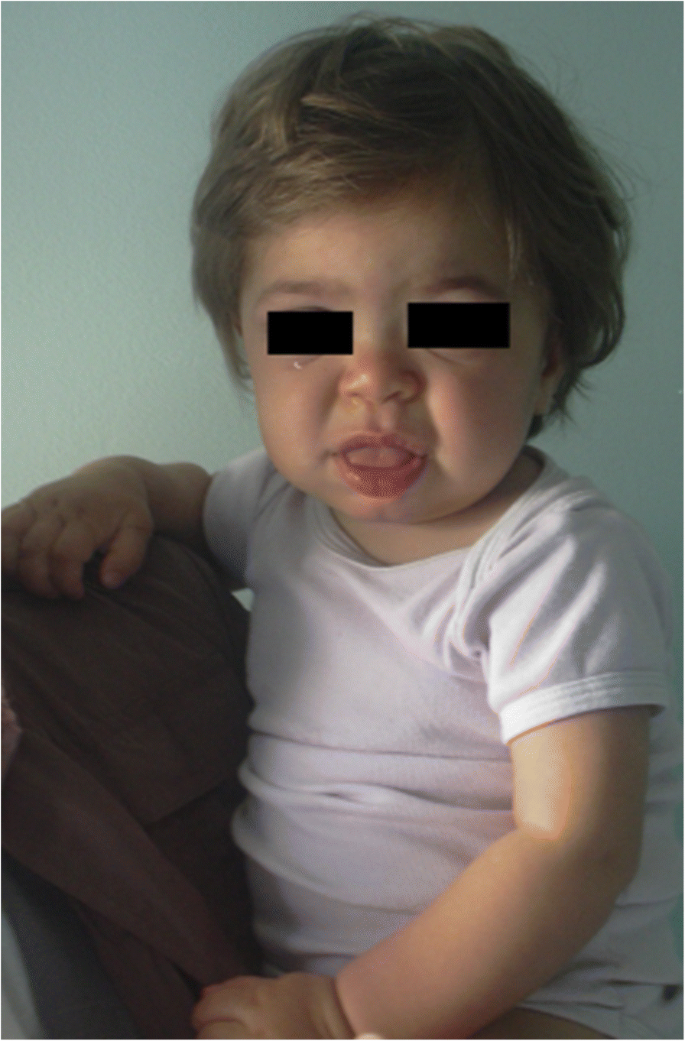
MPS IH bij 1-jarige patiënt. Grove facies: platte neusbrug, macroglossia, frontale bossing
kortom, skeletachtige manifestaties zijn typisch en soms vroegrijp. Grove gezichten en cognitieve vertraging zijn niet vroege prominente tekenen.
Wat zijn de verschillende presentaties van de verschillende typen parlementsleden?
MPS met somatische en cognitieve betrokkenheid
mucopolysaccharidose type I (MPS i; omim #252800) (Fig. 1, 2, 3, 4 en 5) wordt veroorzaakt door een tekort aan de lysosomale hydrolase α-l-iduronidase, wat leidt tot multisystem accumulatie van twee Gag ‘ s: dermatansulfaat (DS) en heparansulfaat (HS) . Op basis van de ernst ervan is MPS I traditioneel onderverdeeld in drie klinische fenotypen; deze vormen vertegenwoordigen echter een continuum van ernst van de ziekte. Hurler syndroom, de meest ernstige vorm, meestal presenteert tijdens het eerste jaar van het leven. De aangetaste kinderen ontwikkelen snel significante cognitieve stoornissen en somatische ziekte in meerdere orgaansystemen, die binnen het eerste decennium zonder behandeling tot de dood leiden. De verzwakte vormen van MPS I, bekend als Hurler-Scheie syndroom en Scheie syndroom, respectievelijk, worden gekenmerkt door latere aanvang van de symptomen, langere levensverwachting, en milde of geen centrale zenuwstelsel (CZS) betrokkenheid . Overerving is autosomaal recessief en in ten minste 50% van de gevallen is fenotype voorspelling mogelijk op basis van genotype, terwijl in de resterende 50% nieuwe mutaties worden gedetecteerd . De prevalentie is ongeveer 1 op de 100.000 levendgeborenen .

MPS IH. een Gibbus in een 9 maanden oude patiënt. B wervelkolom x-ray van de wervelkolom bij een 3-jarige patiënt. c T2-gewogen magnetische resonantiebeeld van de wervelkolom met significante kyfose en vertebrale stenose

MPS IH. X-ray bewijs van dysostose multiplex. verdikking van de ribben bij een 3-jarige patiënt en B heupdysplasie bij een andere patiënt van 18 maanden

MPS IH: een Klauwhand bij een 18 maanden oude patiënt; B hand X-ray.

MPS IH. verdikking van het corticale bot van de schedel en abnormale” J-vormige ” conformatie van de Sella turcica. Dilatatie van de periventriculaire ruimten in B-FLAIR en c T-2 gewogen magnetische resonantiebeelden van een patiënt in de leeftijd van 17 maanden
mucopolysaccharidose type II (MPS II; omim #309900) (Fig. 6), Ook bekend als Hunter-syndroom, is het resultaat van deficiëntie van het enzym iduronaat-2-sulfatase (I2S) met de daaruit voortvloeiende GAG-accumulatie van DS en HS . De prevalentie is ongeveer 1 op 140.000-156.000 mannelijke levendgeborenen . Met name, is dit de enige x-verbonden MPS, en de vrouwelijke dragers zijn gewoonlijk asymptomatisch; nochtans, kunnen zij uitzonderlijk wegens abnormaliteiten van het X-chromosoom, homozygositeit, of scheef-x-inactivering worden beà nvloed . De fenotypische uitdrukking overspant ook een breed spectrum van klinische strengheid. In ernstige gevallen, duidelijke progressieve neurologische betrokkenheid en somatische ziekte naast elkaar bestaan, en de dood treedt meestal in het tweede decennium van het leven. Andere patiënten met minimale of geen neurologische disfunctie kunnen een ernstige somatische betrokkenheid vertonen, terwijl er aan de andere kant van het spectrum patiënten zijn met slechts minimale somatische manifestaties en normale intelligentie die tot in de volwassenheid overleven . Vroege tekenen en symptomen worden gedeeld door de ernstige vormen van MPS I en II.

MPS II. Een 3-jarige patiënt met slechts milde karakteristieke gelaatstrekken
sporadische gevallen van hydrops foetalis worden beschreven in MPS I, hoewel deze kinderen in het algemeen normaal lijken bij de geboorte. De eerste tekenen van MPS I verschijnen meestal tijdens de eerste maanden van het leven, terwijl die in MPS II meestal iets later optreden . Kiely et al. , in een retrospectieve studie bij 55 Hurler patiënten, waargenomen dat respiratoire symptomen, voedingsproblemen, inguinale hernia, en otitis media waren verschenen op een mediane leeftijd ≤ 6 maanden, en kyfose, hartafwijkingen, vergrote hoofdomtrek, hypotonie, organomegalie, cornea vertroebeling, gewrichtsbeperking, en navelhernia verschenen tussen 6 en 12 maanden. Hernia navelstreng is een van de vroegste verschijningskenmerken in MPS I en II, en inguinale hernia worden gemeld bij ongeveer 60% van de patiënten .
een andere veelvoorkomende aanwijzing voor MPS I en II kunnen terugkerende infecties in de bovenste luchtwegen met verhoogde secreties zijn. Frequente otitis, chronische terugkerende rhinitis en aanhoudende loopneus zonder duidelijke infectie zijn niet specifiek, maar kunnen helpen bij het versterken van de diagnostische verdenking indien geassocieerd met meer specifieke tekenen. Opslag van GAG binnen de Oro-farynx leidt tot uitbreiding van de amandelen en adenoïden, en draagt bij aan de bovenste luchtweg complicaties. Een andere frequente bevinding is lawaaierige ademhaling, vooral ‘ s nachts, geassocieerd in de latere stadia met obstructieve slaapapneu. Als gevolg van frequente middenoorinfecties en dysostose van de oogbeentjes van het middenoor, is gehoorverlies frequent, met zowel geleidende als sensorineurale tekorten . Tracheobronchomalacie wordt vaak waargenomen en kan leiden tot acute obstructie of instorting van de luchtwegen, met dit als een van de belangrijkste doodsoorzaken in de daaropvolgende jaren .
adenoïdectomie, tonsillectomie en tympanostomie, samen met herniaherstel, zijn de frequentere en vroege chirurgische ingrepen voor MPS I-en II-patiënten, vaak uitgevoerd voordat de diagnose bekend was (in meer dan 50% van de gevallen) .
recidiverende diarree komt vrij vaak voor in de vroege stadia van MPS I en kan ook voorkomen bij MPS II-patiënten met neurologische betrokkenheid, terwijl het soms voorkomt bij de licht getroffen patiënten .
Gibbusdeformiteit (dorso-lumbale kyfose) (Fig. 2) wordt klinisch duidelijk op een mediane leeftijd van 8.6 maanden tot 1 jaar in MPS I, wat een vrij specifieke en vroegrijpe marker van MPS in het algemeen vertegenwoordigt. Vertebrale lichamen lijken afgeplat en snavelvormig, wat mogelijk leidt tot latere complicaties zoals beknelling van de wervelzenuw, acuut letsel aan de wervelkolom en instabiliteit van het atlanto-occipitale gewricht; daarom moet speciale aandacht worden besteed aan het voorkomen van dislocatie van het atlanto-axiale gewricht tijdens algemene anesthesie en chirurgie. Na de leeftijd van 1 jaar vertegenwoordigen gewrichtscontracturen en genu valgum andere frequente tekenen van gewrichtsstoornis en progressieve skeletdysplasie, die gemakkelijk op de leeftijd van 2 jaar te detecteren zijn bij alle zuigelingen die last hebben van MPS. Al deze skeletachtige manifestaties samen, typisch voor MPS, worden dysostose multiplex genoemd. Verdikking van de ribben is te zien op röntgenfoto ‘ s (Fig. 3a), de lange botten zijn kort met brede schachten en de knieën zijn gevoelig voor valgus-en varus-misvormingen. Phalangeale dysostose en synoviale verdikking leiden tot een karakteristieke klauw hand misvorming (Fig. 4), dat is een ander typisch kenmerk dat vermoeden van de ziekte kan verhogen. Heupdysplasie is vaak de reden waarom deze kinderen voor het eerst kunnen worden gezien door orthopedische specialisten. Meestal is het bekken slecht gevormd, de femurkoppen zijn klein en coxa valga komt vaak voor, met progressieve en slopende heupvervorming (Fig. 3b).
er is een opmerkelijk verschil tussen MPS I en MPS II; bij MPS I neemt de skeletgroei af met de leeftijd van 3 jaar, terwijl MPS II-patiënten overgroei vertonen in de eerste 5-6 jaar , waardoor hun groei daarna wordt vertraagd .
grofheid van de gelaatstrekken (brede neus met uitlopende neusgaten, prominente supraorbitale ruggen, grote afgeronde wangen, dikke lippen, uitpuilende tong) als gevolg van de opslag van Gag ‘ s in de weke delen van het orofaciale gebied en gezichtsbeenderen wordt zichtbaar binnen de eerste 2 jaar, bij een mediane leeftijd van 1,1 jaar voor Hurlerziekte en tussen 18 maanden en 4 jaar in de ernstige vorm van Hunter disease , hoewel subtiele fenotypische veranderingen eerder kunnen worden gedetecteerd (al in de tweede helft van het eerste jaar) door kinderartsen met specifieke ervaring (Fig. 1). Macrocefalie wordt geleidelijk duidelijker, en scafocefalie komt vaak voor (Fig. 5), maar microcefalie is ook mogelijk bij Hurler patiënten . Gezichts-en lichaamshirsutisme zijn altijd duidelijk door de leeftijd van 24 maanden . De huid kan verdikt en inelastisch zijn. Met name ontwikkelen sommige MPS II-patiënten een onderscheidende huidlaesie, die wordt beschreven als ivoorwitte papels (kiezelachtig) op de bovenrug en zijkanten van de bovenarmen, pathognomonisch van het Hunter-syndroom .
cardiomyopathie en progressieve verdikking en stijfheid van de klepbijsluiters bij MPS I-patiënten kunnen leiden tot mitralis-en, minder vaak, aorta-regurgitatie . Een ruis kan worden gedetecteerd in de eerste maanden van het leven en is soms gemeld als het eerste teken van de ziekte . Cardiale betrokkenheid is ook aanwezig bij bijna alle patiënten met MPS II, maar dit begint op ongeveer 5 jaar oud . Klepziekte kan leiden tot rechter en linker ventriculaire hypertrofie en hartfalen.
uitstulping van de buik veroorzaakt door progressieve hepatosplenomegalie is gemakkelijk zichtbaar na de leeftijd van 6 maanden, hoewel opslag van Gag ‘ s in de lever en milt niet leidt tot orgaandisfunctie .
vertroebeling van het hoornvlies komt voor bij bijna alle personen met MPS I op de leeftijd van 15 maanden , wat mogelijk ernstige visusstoornissen kan veroorzaken. Integendeel, cornea vertroebeling is niet een typisch kenmerk van Hunter syndroom, maar spleet-lamp onderzoek kan discrete cornea laesies die het gezichtsvermogen niet beïnvloeden onthullen. Retinale dysfunctie kan worden gedetecteerd, terwijl glaucoom is niet een gemeenschappelijke bevinding.Vroege psychomotorische ontwikkeling wordt over het algemeen als normaal gedetecteerd, maar bij MPS I is ontwikkelingsachterstand meestal duidelijk op de leeftijd van 18 maanden, met een progressieve mentale achteruitgang die leidt tot een ernstige verstandelijke handicap. De taalvaardigheid van deze kinderen is zeer beperkt. Een wereldwijde vertraging van ontwikkelingsmijlpalen in MPS II wordt duidelijk vanaf 2 jaar . Het belangrijkste neurologische kenmerk van de ernstige ziekte van Hunter wordt echter weergegeven door duidelijke gedragsstoornissen, zoals hyperactiviteit, koppigheid en agressiviteit, die doorgaans niet worden waargenomen bij patiënten met het verzwakte fenotype .
mucopolysaccharidose Type VII (MPS VII; omim # 253220), ook bekend als het Sly-syndroom, is een uiterst zeldzame MPS die wordt gekarakteriseerd door een deficiënte activiteit van β-glucuronidase, met lysosomale opslag van chondroïtinesulfaat (CS), DS en HS, wat leidt tot cellulaire en orgaandisfunctie. De eerste patiënt werd beschreven door Sly et al. in 1973 en een totaal van 145 patiënten zijn gemeld in de literatuur tot nu toe.
de klinische presentatie en ziekteprogressie van MPS VII omvat een breed spectrum van ernst, van vroege, ernstige, multisystem manifestaties en overlijden in de eerste maanden, tot een milder fenotype met een latere aanvang, normale of bijna normale intelligentie, en een langere overleving .
fenotype kenmerken van patiënten met MPS VII lijken op die van MPS I en MPS II (korte gestalte, skeletdysplasie, macrocefalie, hypertrofie van het tandvlees, terugkerende oorinfecties, hepatosplenomegalie, hernia ‘ s, betrokkenheid van het hart, verminderde longfunctie en cognitieve stoornissen). Een onverwacht hoog percentage van de beschreven patiënten (41%) heeft een voorgeschiedenis van niet-immuunneonatale hydrops foetalis (NIHF) . Ondanks het prenatale begin van de ziekte bij deze patiënten, overleefden 13 van de 23 de kindertijd met een mild tot gemiddeld verloop in hun late tienerjaren. De aanwezigheid van NIHF voorspelt op zichzelf dus niet de mogelijke ernst van het klinische verloop als de patiënt de kindertijd overleeft. Een ander vroeg teken is macrocrania, terwijl hartklep betrokkenheid en herhaalde infecties van de bovenste en onderste luchtwegen verschijnen in de eerste 2 jaar van het leven. Matige mentale retardatie en progressief gehoorverlies met spraakstoornis zijn ook duidelijk met de tijd.
samengevat hebben de ernstige vormen van MPS I een zeer vroeg multisysteem aanvang (binnen 6 maanden na het leven); MPS II is vergelijkbaar, maar met een latere aanvang, en MPS VII heeft een prenatale aanvang met frequente NIHF en vroege ernstige betrokkenheid van de bovenste en onderste luchtwegen.
MPS met voornamelijk neurologische en cognitieve betrokkenheid
Mucopolysaccharidoses type III (MPS III, ook bekend als het syndroom van Sanfilippo; Fig. 7) heeft een overwegend neurologische presentatie. MPS III is een lysosomale opslagstoornis veroorzaakt door deficiëntie van een van de vier enzymen die betrokken zijn bij het katabolisme van HS. Vier verschillende subtypen bekend zijn—MPS III type A (OMIM #252900), type B (OMIM #252920), type C (OMIM #252930) en type D (OMIM #252940)—elk door een ander enzym deficiëntie: type A, sulfamidase/SGSH gen; type B, alfa-N-acetylglucosaminidase/NAGLU gen; type C, alfa-glucosaminide N-acetyltransferase/HGSNAT gen; type D, N-acetilglucosamina-6-solfato sulfatasi/GNS-gen). Alle vier de typen (A tot en met D) hebben autosomaal recessieve overerving. MPS III is de meest voorkomende MPS met een geschatte prevalentie tussen 0,3 en 4.1 gevallen voor elke 100.000 pasgeborenen, afhankelijk van het subtype en ras .

MPS IIIA. Dezelfde patiënt van 2,5 en 5 jaar. Let op de verschillende gezichtsuitdrukking, die progressie van de neurologische stoornis laat zien
patiënten met het syndroom van Sanfilippo vertonen progressieve cognitieve stoornissen in het tweede en derde levensjaar. In tegenstelling tot de meerderheid van de parlementsleden, zijn somatische kenmerken relatief minder duidelijk, en de betrokkenheid van het CZS is opvallend. Spraakontwikkeling vertraging is meestal het eerste teken dat door de ouders wordt opgemerkt, maar de belangrijkste zorgen kunnen gedragsproblemen zijn (hyperactiviteit, angstig en agressief gedrag, slaapstoornissen), gevolgd door progressieve mentale achteruitgang . Deze symptomen kunnen leiden tot een verkeerde diagnose als gedragsstoornissen, attention deficit hyperactivity disorder (ADHD), en autisme spectrum stoornissen . Epilepsie kan ook aanwezig zijn.
slaapstoornissen, waarvan de incidentie tot 80-90% wordt gemeld, komen vaak voor. Ze bestaan uit moeilijkheden bij het in slaap vallen en frequent nachtelijk ontwaken, met volledige omkering van het dag–nachtritme bij sommige patiënten.
al deze manifestaties zijn zeer moeilijk te beheren en hebben een enorme psychologische impact op de kwaliteit van het leven voor het hele gezin.
hoewel het fenotype bij de vier subtypes zeer vergelijkbaar kan zijn, lijkt het klinische verloop bij Sanfilippo B en C over het algemeen te worden gekenmerkt door een minder ernstig fenotype .
niet-neurologische symptomen zijn gewoonlijk minder uitgesproken bij MPS III dan bij de andere MPS. Terugkerende oor -, neus-en keelinfecties worden waargenomen op jongere leeftijd. Terugkerende otitis, middenoor ossicle defecten, en afwijkingen in het binnenoor kan doofheid veroorzaken. Een overmaat aan dikke afscheidingen en anatomische veranderingen kan obstructie van de luchtwegen veroorzaken. In de eerste levensjaren wordt vaak diarree gemeld, terwijl constipatie vaker voorkomt bij oudere patiënten.
lichamelijk onderzoek kan Grove gelaatstrekken vertonen, hoewel deze minder uitgesproken zijn. Patiënten hebben macrocefalie, brede wenkbrauwen met mediale flaring en synophrys, het haar is meestal droog en grof en hypertrichose komt vaak voor. Bij jongere patiënten worden milde hepatomegalie en navel-en liesbreuken vaak waargenomen. Zeldzame, en vaak milde, contracturen worden meestal gevonden in de ellebogen. Bij ongeveer de helft van de patiënten ouder dan 12 jaar wordt de groei beïnvloed .
samengevat heeft de ziekte van MPS III of de ziekte van Sanfilippo een prominente neurologische betrokkenheid met zeer milde somatische symptomen. Peuters van 2-3 jaar oud die hyperactiviteit, ADHD en agressief gedrag ontwikkelen, kunnen ervan worden verdacht MPS III te hebben.
MPS met prevalente somatische betrokkenheid
MPS IV en MPS VI aanwezig zijn met alleen somatische betrokkenheid. Dit zijn progressieve condities die intellectuele vermogens sparen en voornamelijk het skelet (MPS IVA) of alle organen/systemen van het lichaam (MPS VI) beïnvloeden. Overerving is autosomaal recessief. De snelheid waarmee de symptomen verergeren varieert tussen de getroffen individuen.Mucopolysaccharidose IV (MPS IV, ook bekend als morquiosyndroom; Fig. 8) presenteert zich als twee genetisch verschillende aandoeningen, elk met een verschillende enzymdeficiëntie: MPS IVA (morquio a-syndroom; omim #253000) als gevolg van deficiëntie van n-acetylgalactosamine-6-sulfatase (GALNS-gen), resulterend in accumulatie van keratan-sulfaat (KS) en CS ; en MPS IVB (morquio B-syndroom; omim #253010) als gevolg van deficiëntie van bèta-galactosidase-activiteit die leidt tot urine-KS en oligosaccharide-excretie zoals bij GM1-patiënten. MPS IVB is inderdaad allelisch voor de verschillende vormen van GM1-gangliosidose, maar mist de psychomotorische verslechtering die bij GM1 gangliosidose is waargenomen .

MPS IVA. een 4-jarige patiënt met B antero-posterior en c laterale röntgenfoto ‘s met” paddle-vormige ” ribben en afgeplatte en afgeronde wervels met een typisch “anterior snavel” aspect
patiënten met ernstige MPS IVA zonder aantoonbare enzymactiviteit vertonen meestal hun eerste symptomen tijdens het eerste levensjaar, hoewel ze zelden worden gediagnosticeerd vóór de leeftijd van 4-5 jaar. Informatie over de natuurlijke geschiedenis van Morquio a-patiënten is voornamelijk afkomstig uit twee uitgebreide verzamelingen van gegevens . De verminderde groeisnelheid begint op de leeftijd van ongeveer 18 maanden en de groei stopt op de leeftijd van ongeveer 7 of 8 jaar. In tegenstelling tot de andere MPS, Morquio a gewrichten zijn meestal lax en zeer flexibel (hypermobiel), maar sommige gewrichten (meestal de heupen en schouders) kunnen een beperkt bereik van de beweging. Patiënten met een dergelijke aantasting van het skelet kunnen een diagnose krijgen van, of een evaluatie ondergaan voor, spondyloepifysaire dysplasie, pseudoachondroplasie, meervoudige epifysaire dysplasie of bilaterale ziekte van Legg-Calvé-Perthes .
een constant kenmerk is odontoïde hypoplasie; Zo kan dislocatie van het atlanto-occipitale gewricht optreden waardoor compressie en schade aan de bulb en het ruggenmerg, resulterend in verlamming of zelfs de dood als cervicale stabilisatie niet vroegtijdig wordt uitgevoerd.
MPS IVB patiënten hebben een zeer vergelijkbare klinische manifestatie, maar met een minder prominente korte gestalte. Ze hebben mild Grove gelaatstrekken, gehoorverlies, hoornvliesopaciteiten, hartklepziekte en frequente infecties van de bovenste luchtwegen. Ze kunnen ook aanwezig zijn met inguinale hernia en milde hepatomegalie. Skeletdysplastische kenmerken omvatten platyspondylie, odontoïde hypoplasie met mogelijke cervicale subluxatie, kypho-scoliose, coxa valga en vernauwde iliacale vleugels .
mucopolysaccharidose VI (MPS VI, ook bekend als maroteaux-Lamy syndroom; Omim # 253200; Fig. 9) is te wijten aan pathogene mutaties in het arylsulfatase B (ARSB) gen op chromosoom 5. Als gevolg hiervan heeft een verminderde of afwezige activiteit van het enzym arylsulfatase B (ASB) (n-acetylgalactosamine 4-sulfatase) een negatieve invloed op de afbraak van DS bij het bepalen van de cellulaire accumulatie .

MPS VI. een 2-jarige jongen met duidelijke grof gezicht en skeletdysostose
patiënten zonder aantoonbare enzymactiviteit hebben de ernstige vorm van MPS VI en vertonen symptomen in de vroege kindertijd. Hun somatische fenotype is vergelijkbaar met het hurlersyndroom. In de meeste gevallen, op 2 of 3 jaar oud, ernstige en progressieve skeletschade, genoemd dysostose multiplex, wordt duidelijk. Deze skeletdysplasie omvat heupdysplasie met dysplastische femurkop, abnormale wervellichamen, onregelmatige sleutelbenen, hypoplastische distale ellepijp en radius, en dysplastische en korte metacarpale botten; carpale en tarsale botten zijn hypoplastisch en hebben een onregelmatig profiel. De groeisnelheid vertraagt vaak na het eerste levensjaar, met een uiteindelijke hoogte over het algemeen lager dan 120 cm. Net als bij andere parlementsleden is een vroeg teken Grove gelaatstrekken. Daarnaast hebben patiënten een typische uitstekende buik, hepatomegalie, navel en/of inguinale hernia, en betrokkenheid van het hart in de vroege kindertijd. Hoewel er geen primaire verstandelijke beperking aanwezig is, kan hydrocephalus worden gezien bij sommige patiënten met de daaruit voortvloeiende intracraniële hypertensie en papilledeem. Pectus carinatum, stijve en gecontracteerde gewrichten, scoliose en kyfose, en cervicale ruggenmerg compressie verergeren met de tijd .
samengevat vertonen MPS IV en VI geen betrokkenheid bij het CZS. MPS IV is een unieke MPS in het tonen van gezamenlijke laxiteit, terwijl ernstige MPS VI aanvang vergelijkbaar is met die waargenomen bij MPS I.