Article
Yosivan Gomez Lima1*, Marcel Catao Ferreira dos Santos1, Jullian Tamara Araujo de Melo Campos2
1 Wydział Medycyny Klinicznej, dyscypliny endokrynologicznej i metabologii. Hospital Universitário Onofre Lopes, Universidade Federal do Rio Grande Do Norte (UFRN), Natal, RN, Brazylia
2faculty of Health Sciences of Trairi, Federal University of Rio Grande Do North (UFRN), Natal, RN, Brazylia
Streszczenie
wrodzona uogólniona lipodystrofia (CGL) jest rzadką i ciężką chorobą autosomalną.choroba recesywna. Pacjenci są wadliwi w przechowywaniu tkanki tłuszczowej i w konsekwencji odkładają tłuszcz w tkankach pozamacicznych, głównie w wątrobie, i mogą rozwinąć marskość wątroby. Insulinooporność jest typowym odkryciem, powodującym cukrzycę, która wymaga dużych dziennych dawek insuliny. W stanie Rio Grande Do Norte w Brazylii mamy jedną z największych kohort pacjentów z CGL. W tym artykule dokonujemy przeglądu patofizjologii, obrazu klinicznego i leczenia tej choroby.
wprowadzenie
cukrzyca typu 2 jest światowym problemem zdrowotnym i zwykle wynika z nadmiernej masy ciała i zwiększonego tłuszczu trzewnego, powodując obwodową insulinooporność i niezdolność trzustki do uwalniania insuliny w celu skompensowania tej oporności. Inne mniej powszechne typy cukrzycy występują z powodu specyficznych mutacji genetycznych, takich jak wrodzona uogólniona lipodystrofia (CGL), znana również jako wrodzona lipodystrofia Berardinellego-Seipa (Bscl). CGL jest chorobą autosomalną recesywną, która dzieli się na cztery typy, oparte na mutacji genowej. Zmienione geny odgrywają istotne funkcje dla tworzenia adipocytów, produkcji lipidów i właściwego przechowywania wewnątrz adipocytów. Mutacje zmniejszają tkankę tłuszczową, co powoduje odkładanie się tłuszczu w miejscach pozamacicznych, powodując wątrobę tłuszczową, zmieniony metabolizm węglowodanów, ciężką oporność na insulinę z hiperinsulinemią i cechami akromegaloidowymi oraz dyslipidemia1-3. Zespół CGL ma około 500 przypadków zgłoszonych na świecie. W Brazylii, w stanie Rio Grande Do Norte (RN), zdiagnozowano, leczono i śledziliśmy 54 przypadki w ciągu ostatnich 20 lat4, 5. W badaniu opisowym wykorzystującym dane wtórne oszacowaliśmy łącznie 103 pacjentów w RN6. Wskazuje to na znacznie większą częstość występowania niż w literaturze (1: 1 mln) 7.
tworzenie i przechowywanie Triacyloglicerolu w kroplach lipidowych
biosynteza trójglicerydów i fosfolipidów (Fig. 1A) rozpoczyna się od acylotransferazy glicerolu-3-fosforanowej (GPAT) acylującej w glicerolu-3-fosforanie w pozycji 1, tworząc 1-acyloglicerolu-3-fosforan (kwas lizofosfatydowy). Następnie następuje kolejny etap acylacji w pozycji drugiej przez enzym AGPAT (1-Acyloglicerol-3-fosforan acylotransferaza), pochodzący z 1,2-Diacyloglicerol-3-fosforanu (kwas fosfatydowy). Jest kluczowym etapem pośrednim w szlaku biosyntezy trójglicerydów i fosfoglicerydów. Istnieje 11 izoform enzymów AGPAT, kodowanych przez różne geny4. AGPAT1 i AGPAT2 są najszerzej badane. AGPAT1 jest obecny na wysokim poziomie w jądrach, trzustce i, w mniejszym stopniu, w tkance tłuszczowej i innych tkankach, takich jak serce, łożysko, mózg, płuca, podczas gdy AGPAT2 jest obfity w tkankę tłuszczową. W kolejnych etapach, enzym cytozolowy fosfataza kwasu fosfatydowego (PAP lub lipin) wytwarza 1,2-diacyloglicerol, a acylotransferaza 1,2-diacyloglicerol (DGAT) tworzy triacyloglicerol4. Kwas fosfatydowy i diacyloglicerol mogą również pochodzić z innych fosfolipidów, takich jak kardiolipina, fosfatydyloinozytol i Fosfatydylocholina.
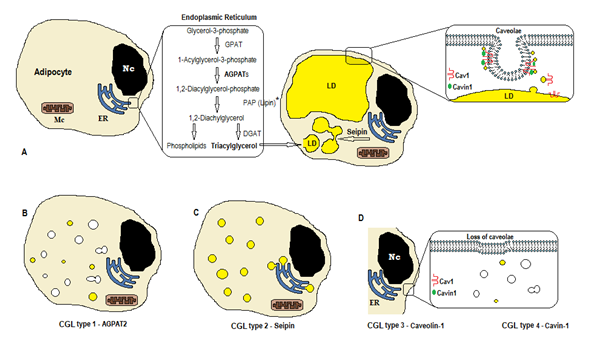
Rysunek 1. Schemat syntezy trójglicerydów według typów CGL. A) normalna synteza i przechowywanie triacyloglicerolu (TAG) w adipocytie. B) mutacja AGPAT2 zmniejsza produkcję znaczników (niektóre są nadal syntetyzowane pod wpływem stymulacji innych Agpatów). (C) mutacja genu seipin zmniejsza syntezę tagów i tworzenie kropel lipidowych (LD) i fuzję. D) Caveolin-1 i Cavin-1 są wymagane do tworzenia i stabilizacji caveolae. Mutacja w CAV1 (typ 3) lub CAVIN1 (Typ 4)może spowodować utratę caveolae w błonie. NC, nucleus. Retikulum endoplazmatyczne. Mc, mitochondria. * Lipina jest enzymem cytozolowym zakotwiczonym przez seipin w ER.
reakcje te występują w retikulum endoplazmatycznym adipocytów (ER), gdzie postępująca kumulacja trójglicerydów powoduje powstawanie małych kropelek lipidowych (LD)8. Produktem genu BSCL2 jest białko transbłonowe zwane seipin, które powoduje fuzję małego LD, pochodzącego z dużego LD. Seipin znajduje się w ER i koncentruje się na skrzyżowaniu z powstającym LD, ułatwiając przepływ lipidów między ER i LD i włączenie triglicerydów do LD9. Seipin może również działać jako kotwica ER dla enzymu cytozolowego Lipiny 1. Oprócz tego, że seipin jest niezbędny do fuzji kropel lipidowych, wielkości i morfologii, jest również niezbędny do adipogenezy (poprzez interakcję z lipiną 1) i komórkowymi trójglicerydami lipolizy10, 11. Niedobór seipin utrudnia różnicowanie pre-adipocytów do adipocytów i wpływa na ostateczną dojrzałość9, jak wykazano w badaniach na mezenchymalnych komórkach macierzystych z bscl212. Tkanki beztłuszczowe również wyrażają seipin, a inne funkcje mają być określone.
w adipocytach caveolae, które są wyspecjalizowanymi inwaginacjami membranowymi o długości 50-100 nm, stanowią 20% powierzchni błony plazmatycznej, co czyni adipocyty komórkami o najwyższej gęstości caveolae13. Tworzenie kropel lipidowych wymaga białka błonowego (Caveolin-główny składnik błon caveolae)i białka cytoplazmatycznego (Cavin-1) 14. Geny CAV1, CAV2 i CAV3 kodują trzy formy caveolinu o podobnych strukturach (odpowiednio Caveolin-1, Caveolin-2 i Caveolin-3). Caveolin-1 i Caveolin-2 są obecne w adipocytach, fibroblastach i komórkach śródbłonka, a Caveolin-3 jest obecny tylko w mięśniu szkieletowym i sercowym13, 15. Caveolin-1 jest najważniejszy i najbardziej przebadany. Wyraża się w dwóch różnych izoformach (1a i 1b). Caveolin – 1 translokuje się z błony plazmatycznej do kropli lipidowych, co jest niezbędne do handlu lipidami i metabolizmu16. Krople lipidowe przechowują trójglicerydy po karmieniu, a cząsteczki te są hydrolizowane do kwasu tłuszczowego i uwalniane podczas postu; mechanizm ten może być regulowany przez Caveolin-116. Niedobór caveoliny – 1 zwiększa również podatność na śmierć komórek przez autofagię17.
Gen CAVIN1 koduje białko cytoplazmatyczne zwane białkiem związanym z caveolae 1 (Cavin-1)14, 16, które jest obowiązkowe do tworzenia i stabilizacji caveolae. Cavin – 1 ulega ekspresji w adipocytach, komórkach mięśniowych i innych komórkach, a także jest niezbędny w przekazywaniu sygnałów pochodzących z caveolae14, 18. Nokaut genu CAV1 powoduje brak caveolae w komórkach nie-mięśniowych, podczas gdy nokaut CAVIN1 powoduje brak caveolae we wszystkich tkankach, w tym mięśnie14. Brak caveolae może wpływać na regulację lipolizy, strumienia kwasów tłuszczowych, syntezy trójglicerydów i sygnałów innych szlaków.
rodzaje CGL
na podstawie wykrywalnych zmian genetycznych opisano cztery typy. Typy 1 i 2 są odpowiedzialne za ponad 95% przypadków, a typ 2 ma bardziej dotknięty fenotyp. Zgłoszono tylko jeden przypadek typu 3 i około 30 przypadków typu 44.
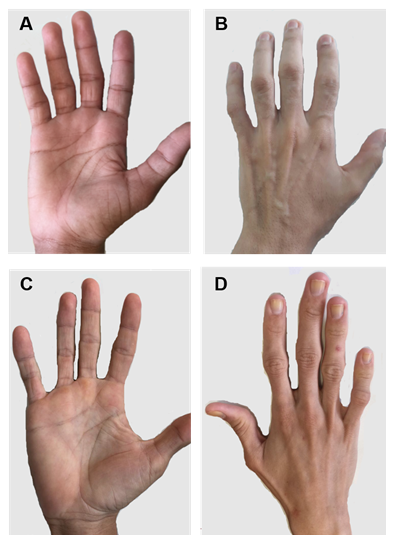
Rysunek 2. Ręce pacjentów z typami CGL 1 i 2. A) i B) przedni i tylny widok dłoni pacjentów typu 1. Najwyraźniej normalne ręce, ponieważ nadal istnieje mechaniczna tkanka tłuszczowa. C) I D) widok przedni i tylny dłoni pacjentów typu 2. Nasilenie choroby jest większe, a brak tłuszczu jest widoczny i łatwo zauważalny.
CGL Typ 1. W 1999, Garg et al. opisał mutację pacjentów na chromosomie 9q34, a trzy lata później Agarwal i wsp. wykazywał AGPAT2 jako enzym dotknięty tą mutacją2, 19. Z powodu mutacji tego AGPAT2, Żadna lub minimalna produkcja triacyloglicerolu odbywa się pod wpływem bodźców innych izoform. Fenotyp myszy nokautującej AGPAT2 jest podobny do ludzkiego z typem CGL, co potwierdza rolę tego enzymu w patofizjologii20, 21.
CGL Typ 2. Magre et al. jako pierwsi zidentyfikowali mutację w genie seipin (chromosom 11q13) 3. Mutacje (głównie nonsensowne) genu seipin (bscl2) wytwarzają obcięte białko i mogą wpływać na metabolizm lipidów za pomocą różnych mechanizmów: a) zmniejszenie stabilności seipin; B) zmniejszenie zdolności wiązania Lipiny 1; i c) Brak oligomeryzacji i lokalizacji wyłącznie na błonie ER11. Niektóre komórki są nadal w stanie wytwarzać triacyloglicerol i małe kropelki lipidowe, ale duże kropelki lipidowe są nieobecne z powodu utraty zdolności fuzji tych małych kropelek lipidowych. Istnieje również niepowodzenie w ekspresji czynników adipogennych, takich jak aktywowany przez proliferator peroksysomów receptor gamma (PPARG), a także adiponektyna i białko wiążące kwas tłuszczowy adipocytów (FABP4)11, 16. Niedobór Seipin upośledza adipogenezę, zwiększa lipolizę i zapobiega gromadzeniu się trójglicerydów w adipocytach.
CGL typ 3. Ten typ został niedawno opisany u pacjenta, który pomimo fenotypu CGL nie miał mutacji w genach AGPAT2 lub BSCL222. Myszy z mutacją Cav1 są odporne na otyłość wywołaną dietą i mają insulinooporność, hipertriglicerydemię, zmniejszoną adiponektynę, zmniejszoną masę tłuszczową i małe adipocyty16. Po wyborze genów kandydujących na podstawie badań na myszach, Kim et al. potwierdzono obecność mutacji nonsensownej w genie cav1 (CAV1) na chromosomie 7q3122.
CGL Typ 4. W tym rzadkim typie genem jest CAVIN1, który koduje białko Cavin-1. U ludzi obserwowano go u pacjentów z uogólnioną wrodzoną lipodystrofią i dystrofią mięśniową15, 23.
ostatnio opisano również mutacje w genach PCYT1A i PPARG powodujące lipodystrofię24, 25.
cechy kliniczne
u pacjentów z CGL zwykle występują akromegaloidy, akantoza czarna, febomegalia, hepatomegalia i przerost mięśni5, 26, 27. Niektórzy autorzy powołują się na przepuklinę pępkową jako kliniczne odkrycie syndromu26. Oceniliśmy częstość występowania tego zjawiska u naszych pacjentów i żaden z nich nie przedstawił tej zmiany28. W rzeczywistości brak okołomięśniowej tkanki tłuszczowej powoduje wysunięcie blizny pępowinowej, co może być błędnie zdiagnozowane jako przepuklina28, 29.
gdy adipocyty nie są w stanie odpowiednio przechowywać tłuszczu, gromadzi się on w innych tkankach, takich jak wątroba i mięśnie, powodując ciężką insulinooporność. Densytometria kości (DXA)może wykazywać prawidłową lub wysoką gęstość mineralną kości30 i zmniejszenie całkowitej tkanki tłuszczowej (zwykle poniżej 6%) 27. W wyniku niskiej zawartości tłuszczu w organizmie, adiponektyna w surowicy i leptyna są zbyt niskie27. Ponieważ leptyna jest niezbędna w kontrolowaniu głodu, pacjenci ci zazwyczaj mają hiperfagię, która jest łatwo widoczna od dzieciństwa. Adiponektyna odgrywa ważną rolę jako środek uczulający na insulinę, a jej brak pogarsza oporność na insulinę. Pomimo tego, początkowo glukoza i glikowana hemoglobina są normalne kosztem bardzo wysokiego poziomu insuliny. Cukrzyca zwykle rozpoczyna się w okresie dojrzewania; w naszej serii średni wiek zachorowania wynosił 15,8±7,1 lat27. Początkowo są one kontrolowane za pomocą leków doustnych, które wymagają dużych dawek insuliny w ciągu kilku lat27. Nadciśnienie tętnicze występuje u jednej trzeciej pacjentów27.
istnieją pewne specyficzne cechy kliniczne każdego typu CGL. U pacjentów z typem 1 nadal występuje mechaniczny tłuszcz tłuszczowy, zwłaszcza w dłoniach,podeszwach, oczodołach, okolicach stawowych31. Natomiast pacjenci typu 2 wykazują brak metabolicznych i mechanicznych tkanek tłuszczowych. Seipin jest silnie wyrażony w mózgu i móżdżku, a także bierze udział w regulacji funkcji nerwowych. Ponad połowa pacjentów typu 2 ma pewne upośledzenie funkcji poznawczych1, 8. Typy 3 i 4 mają zachowanie tłuszczu mechanicznego i szpiku kostnego, a Typ 4 ma osłabienie mięśni związane z wysoką kinazą kreatynową w surowicy i niestabilnością kręgosłupa 15.
istnieją również cechy kliniczne specyficzne dla płci. Często występują policystyczne jajniki i brak miesiączki32. Cykle miesiączkowe zwykle wracają do normy po zastosowaniu metreleptyny, prawdopodobnie z powodu poprawy wrażliwości na insulinę i przywrócenia pulsacji LH32. Mężczyźni Typu 2 mogą mieć teratozoospermię z powodu braku seipin w komórkach zarodkowych33.
hipertriglicerydemia występuje od pierwszych lat życia i może powodować ostre zapalenie trzustki. HDL jest zwykle mniejsza niż 30 mg/dL. Zwiększenie aktywności enzymów wątrobowych jest również wczesnym stwierdzeniem i pochodzi z odkładania się tłuszczu w wątrobie. Postępujące zmniejszenie liczby płytek krwi w surowicy wskazuje na nasilenie choroby wątroby i prawdopodobną marskość marskości34.
ponieważ Cavin-1 jest obecny w komórkach mięśniowych, u pacjentów z typem 4 występuje łagodne osłabienie mięśni i zwiększenie kinazy kreatynowej15.
średnia długość życia, głównie w typie 2, jest znacznie zmniejszona, a śmierć nierzadko występuje przed ukończeniem 30 lat (osobiste doświadczenie oparte na 20 pacjentach, którzy zmarli w ciągu ostatnich 19 lat). Przyczyny zgonu są związane z cukrzycą (niewydolność nerek, nagła śmierć), wątrobą (marskość wątroby, krwawienie z przewodu pokarmowego) lub infekcjami.
Diagnostyka i leczenie
diagnoza CGL opiera się na danych klinicznych: cechy akromegaloidów, rogowacenie czarnuszki, zmniejszenie całkowitej tkanki tłuszczowej, przerost mięśni i występ blizny pępowinowej. Ponadto dane laboratoryjne mogą wykazać cukrzycę z ciężką insulinoopornością i hipertriglicerydemią. Badania obrazowe mogą pomóc zidentyfikować ektopowe złogi tłuszczu głównie w wątrobie i trzustce (stłuszczenie wątroby z hepatomegalią i stłuszczeniem trzustki). DXA może potwierdzić niski poziom tkanki tłuszczowej i wysoką gęstość kości30.
leczenie CGL polega na ścisłej kontroli diety ze zmniejszeniem spożycia tłuszczów, głównie trójglicerydów i żywności o wysokim indeksie glikemicznym, w celu zapobiegania chorobom współistniejącym i ich kontrolowania29. Jednak idealna dieta jest trudnym celem do osiągnięcia z powodu zwiększonego apetytu i surowych ograniczeń zalecanych. Należy również zachęcać do aktywności fizycznej w celu poprawy kontroli chorób współistniejących, z wyjątkiem pacjentów z przeciwwskazaniami, takimi jak ciężka kardiomiopatia 29.
jeśli chodzi o leczenie farmakologiczne, pacjenci ci mogą być leczeni zwykłymi lekami na cukrzycę, nadciśnienie i wytyczne dotyczące dyslipidemii. Pierwszym wyborem w leczeniu cukrzycy i insulinooporności jest metformina, ale zwykle nie wystarcza. W przeciwieństwie do leczenia częściowej lipodystrofii, tiazolidynodiony należy stosować z ostrożnością29. Stosuje się inne doustne leki przeciwcukrzycowe, ale nie były one specjalnie badane u pacjentów z CGL. Istnieją dane dotyczące zwierząt sugerujące, że stosowanie inhibitorów SGLT2 (dapagliflozyny) może przynieść korzyści w zapobieganiu kardiomiopatii35; konieczne są badania, aby to potwierdzić u ludzi. Wraz z postępem choroby i wystąpieniem ciężkiej insulinooporności potrzebne są wysokie dzienne dawki insuliny. Brak podskórnej tkanki tłuszczowej stanowi problem w podawaniu dużych dawek insuliny. Może być wymagana bardziej stężona insulina (u-300 lub U500) 36. U tych pacjentów występuje ciężka dyslipidemia, głównie ze względu na wzrost trójglicerydów i niski poziom HDL, a zatem stosowanie fibratu jest czasami konieczne w celu zapobiegania ostremu zapaleniu trzustki. Ponadto, ze względu na wysokie ryzyko sercowo-naczyniowe u tych pacjentów, należy rozważyć interwencję ze statynami, a cele LDL lub nie-HDL powinny być ściśle określone 29.
codzienne zastrzyki metreleptyny powodują znaczne zmniejszenie apetytu i przynoszą korzyści poprzez obniżenie glikemii, trójglicerydemii i enzymów wątrobowych. Zauważalne jest, zwłaszcza u dzieci, zmniejszenie obwodu brzucha, prawdopodobnie z powodu zmniejszenia hepatomegalii.
wniosek
CGL jest rzadką i ciężką chorobą, która może wystąpić z cukrzycą (Zwykle wymagającą dużych dawek insuliny) i przedwczesną śmiercią. Fenotyp pacjenta jest dość charakterystyczny, wymagający jednak znajomości zespołu przez pracowników służby zdrowia do postawienia wczesnej diagnozy. Metreleptyna wydaje się być jedynym lekiem w tej chwili, który może zmienić naturalną historię choroby.
konflikt interesów: brak.
- Nolis T. badając patofizjologię bardziej powszechnych genetycznych i nabytych lipodystrofii. Journal of human genetics. 2014 Jan; 59(1): 16-23.
- Agarwal AK, Arioglu E, De Almeida s, et al. AGPAT2 ulega mutacji w wrodzonej uogólnionej lipodystrofii związanej z chromosomem 9q34. Nat Genet. 2002 May; 31(1): 21-3.
- Magre J, Delepine m, Khallouf E, et al. Identyfikacja genu zmienionego w wrodzonej lipodystrofii Berardinellego-Seipa na chromosomie 11q13. Genetyka natury. 2001 Aug; 28 (4): 365-70.
- Patni N, Garg A. Wrodzona uogólniona lipodystrofia-nowe spojrzenie na zaburzenia metaboliczne. Natura opinie Endokrynologia. 2015 Sep; 11(9): 522-34.
- Garg A. nabył i odziedziczył lipodystrofie. The New England journal of medicine. 2004 Mar 18; 350(12): 1220-34.
- de Azevedo Medeiros LB, Candido Dantas VK, Craveiro Sarmento AS, et al. Wysoka częstość występowania wrodzonej lipodystrofii Berardinellego-Seipa w stanie Rio Grande Do Norte w północno-wschodniej Brazylii. Diabetol Metab Syndr. 2017; 9: 80.
- Chiquette E, Oral EA, Garg a i in. Szacowanie częstości występowania uogólnionej i częściowej lipodystrofii: odkrycia i wyzwania. Cukrzyca, zespół metaboliczny i otyłość: cele i terapia. 2017: 375-83.
- Wee K, Yang w, Sugii S, et al. W kierunku mechanistycznego zrozumienia lipodystrofii i funkcji seipina. Raporty biologiczne. 2014; 34(5).
- Dollet L, Magre J, Cariou B, et al. Funkcja seipin: nowe spostrzeżenia z modeli myszy knockout bscl2 / seipin. Biochimie. 2014 Jan; 96: 166-72.
- Sim MF, Dennis RJ, Aubry EM, et al. Ludzkie białko lipodystrofii seipin jest adapterem membranowym ER dla adipogennej fosfatazy PA lipin 1. Metabolizm molekularny. 2012; 2(1): 38-46.
- Sim MF, Talukder MM, Dennis RJ, et al. Analiza naturalnie występujących mutacji w ludzkim białku lipodystrofii seipin ujawnia wiele potencjalnych mechanizmów patogennych. Diabetologia. 2013 Nov; 56(11): 2498-506.
- Payne VA, Grimsey N, Tuthill A, et al. Ludzki gen lipodystrofii BSCL2 / seipin może być niezbędny do prawidłowego różnicowania adipocytów. Cukrzyca. 2008 Aug; 57(8): 2055-60.
- Cohen AW, Hnasko R, Schubert W, et al. Rola caveolae i caveolins w zdrowiu i chorobie. Przeglądy fizjologiczne. 2004 Oct; 84(4): 1341-79.
- Pilch PF, Liu L. jaskinie tłuszczowe: caveolae, handel lipidami i metabolizm lipidów w adipocytach. Trendy w endokrynologii i metabolizmie: TEM. 2011 Aug; 22 (8): 318-24.
- Hayashi YK, Matsuda C, Ogawa m, et al. Ludzkie mutacje PTRF powodują wtórny niedobór caveolin, co prowadzi do dystrofii mięśniowej z uogólnioną lipodystrofią. J Clin Invest. 2009 Sep; 119(9): 2623-33.
- Parton RG, del Pozo MA. Caveolae jako czujniki membrany plazmowej, ochraniacze i organizery. Nature reviews Molecular cell biology. 2013 Feb; 14 (2): 98-112.
- Le Lay S, Briand N, Blouin CM, et al. Model lipoatroficzny caveolin-1 z niedoborem myszy ujawnia autofagię w dojrzałych adipocytach. Autofagia. 2010 Aug; 6 (6): 754-63.
- Delecja Cavin / PTRF powoduje globalną utratę caveolae, dyslipidemii i nietolerancji glukozy. Metabolizm komórkowy. 2008 Oct; 8(4): 310-7.
- Garg A, Wilson R, Barnes R, et al. Gen wrodzonej uogólnionej lipodystrofii mapuje ludzki chromosom 9q34. The Journal of clinical endocrinology and metabolism. 1999 Sep; 84(9): 3390-4.
- Vogel P, Read R, Hansen G, et al. Patologia wrodzonej uogólnionej lipodystrofii u myszy Agpat2 -/ -. Patologia weterynaryjna. 2011 May; 48 (3): 642-54.
- Cortes VA, Curtis DE, Sukumaran S, et al. Molekularne mechanizmy stłuszczenia wątroby i insulinooporności w mysim modelu wrodzonej lipodystrofii uogólnionej z niedoborem AGPAT2. Metabolizm komórkowy. 2009 Feb; 9(2): 165-76.
- Kim CA, Delepine m, Boutet E, et al. Skojarzenie homozygotycznej mutacji caveolin-1 z wrodzoną lipodystrofią Berardinellego-Seipa. J Clin Endocrinol Metab. 2008 Apr; 93(4): 1129-34.
- Śmiertelna arytmia serca i zespół długiego QT w nowej postaci wrodzonej uogólnionej lipodystrofii z falowaniem mięśni (CGL4) spowodowanej mutacjami PTRF-CAVIN. PLoS genetics. 2010 Mar 12; 6 (3): e1000874.
- Mutacje zaburzające szlak fosfatydylocholiny u ludzi z wrodzoną lipodystrofią i stłuszczeniem wątroby. Proc Natl Acad Sci U S A. 2014 Jun 17; 111 (24): 8901-6.
- Dyment DA, Gibson WT, Huang L, et al. Mutacje bialleliczne w PPARG powodują wrodzoną, uogólnioną lipodystrofię podobną do zespołu Berardinellego-Seipa. Eur J Med Genet. 2014 Sep; 57 (9): 524-6.
- Garg A. przegląd kliniczny#: Lipodystrofie: genetyczne i nabyte zaburzenia tkanki tłuszczowej. The Journal of clinical endocrinology and metabolism. 2011 Nov; 96 (11): 3313-25.
- Lima JG, Nobrega LH, de Lima NN, et al. Dane kliniczne i laboratoryjne dużej serii pacjentów z wrodzoną uogólnioną lipodystrofią. Diabetol Metab Syndr. 2016; 8: 23.
- Lima GJ, Lima NN, Oliveira CF, et al. Przepuklina pępkowa u pacjentów z zespołem Berardinelliseip: czy to naprawdę przepuklina. J Clin Mol Endocrinol. 2015; 1(1): 3.
- Brown RJ, Araujo-Vilar D, Cheung PT, et al. Diagnostyka i Postępowanie w zespołach lipodystrofii: wytyczne dotyczące praktyki wielu społeczeństw. J Clin Endocrinol Metab. 2016 Dec; 101(12): 4500-11.
- Lima JG, Nobrega LH, Lima NN, et al. Gęstość kości u pacjentów z wrodzoną lipodystrofią Berardinellego-Seipa jest większa w miejscach beleczkowatych i u pacjentów typu 2. J Clin Densitom. 2016 Listopad 25.
- Simha V, Garg A. heterogeniczność fenotypowa w rozmieszczeniu tkanki tłuszczowej u pacjentów z wrodzoną uogólnioną lipodystrofią spowodowaną mutacjami w genach AGPAT2 lub seipin. J Clin Endocrinol Metab. 2003 Nov; 88 (11): 5433-7.
- Musso C, Cochran E, Javor e, et al. Długotrwały wpływ leczenia rekombinowaną metionylową ludzką leptyną na hiperandrogenizm i funkcje menstruacyjne u kobiet i przysadki mózgowej u mężczyzn i kobiet pacjentów z hipoleptynemią lipodystroficzną. Metabolizm. 2005 Feb; 54(2): 255-63.
- Brak seipin jąder powoduje zespół teratozoospermii u mężczyzn. Proc Natl Acad Sci U S A. 2014 May 13; 111(19): 7054-9.
- Mitchell O, Feldman DM, Diakow m, et al. Patofizjologia małopłytkowości w przewlekłej chorobie wątroby. Hepat Med. 2016; 8: 39-50.
- Joubert M, Jagu B, Montaigne D, et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes. 2017 Apr; 66(4): 1030-40.
- Lima JG, Lima NN, Lima RLM, et al. Glargine U300 Insulin as a Better Option than Degludec U100 to Treat a Congenital Generalized Lipodystrophy Patient. Clin Diabetes Res. 2017; 1(1).