Autozomal recesiv Congenital ihtioza / Actas Dermo-Sifiliogr Xvficas
Introducere
cea mai recentă clasificare consensuală a ihtiozei diferențiază între 2 forme principale: formele nesindromice, care prezintă doar manifestări cutanate, și formele sindromice, care prezintă manifestări și în alte organe (Tabelul 1).1 dintre formele nesindromice, se identifică 4 grupe: ihtioză comună, ihtioză congenitală autosomală recesivă (ARCIs), ihtioză keratinopatică și alte ihtioză mai puțin frecvente.În mod tradițional, grupul de ARCIs a fost împărțit în 2 tulburări, ihtioză lamelară (LI) și eritrodermie ihtiosiformă congenitală (CIE). În noua clasificare, ihtioza harlequin (HI) a fost adăugată la acest grup1 deoarece mutațiile inactivatoare ale genei ABCA12 au fost identificate ca fiind responsabile pentru această tulburare,2,3 în timp ce mutațiile nonsens din aceeași genă pot da naștere fenotipului LI4 sau CIE5,6. Alte variante mai puțin frecvente incluse în grupul de ARCIs sunt auto-vindecarea collodion baby (SHCB), acral SHCB și ihtioza costumului de baie.7-9
Clasificarea consensuală bazată pe caracteristicile clinice ale Ihtiozei1.
| forme Nedromice | forme sindromice |
| IchthyosesIchthyosis vulgarisRecessive X-linked ichthyosis (nonsindromic )ajimforme majorsharlequin ichthyosisLamellar ichthyosisCongenital ichthyosiform eritrodermaminorforme auto-vindecătoare collodion babyAcral auto-healing collodion babyBathing suit ichthyosisKeratinopathic IchthyosesMajor formeepidermolitic Ihtiozisuperficial epidermolitic ihtiozismforme minoreanular epidermolitic ihtyosiscurth-Macklin Ihtiozăautozomal recesiv ihtiozăepidermolitic nevusalte Formecloricrin keratodermaeritrokeratodermia vararabilisindromul cutanatcongenital reticular ihtiosiform eritrodermicsindromul maklick | sindromic Ihtiosrecesiv legat de X ihtioză (sindromică)ihtioză foliculară, alopecie și fotofobie (IFAP) sindromconradi-Scammann-happle (chondrodysplasia punctata tip 2)sindrom autozomal ihtiozătulburări ale pieliisindromul Ethertonichthyosis-hypothrichosis syndromeichthyosis-sclerozant cholangitis syndrometrichothystrophyneurological disorderssj syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, retard mintal, enteropatie, surditate, neuropatie periferică, ihtioză, keratodermă.
sunt disponibile doar date limitate privind epidemiologia ARCIs. În Statele Unite, a fost estimată o prevalență la naștere de 1 la 100 000 de locuitori pentru LI și de 1 la 200 000 de locuitori pentru CIE. Alte studii au raportat o prevalență combinată pentru LI și CIE de 1 la 200 000 până la 300 000 de locuitori.10,11 în unele țări, cum ar fi Norvegia, prevalența estimată este mai mare (1 la 91 000) din cauza mutațiilor fondatorilor.12 descoperirea a 1 sau a mai multor mutații recurente într-o populație poate fi cauzată de faptul că mutația a avut loc la un moment dat în istorie și a fost apoi transmisă din generație în generație (mutație fondatoare) sau pentru că regiunea genomului în care se găsește mutația are o secvență ADN susceptibilă la mutație (hotspot de mutație). În Spania, prevalența estimată a ARCI este de 1 la 138 000 în populația generală și de 1 la 61 700 în rândul copiilor cu vârsta sub 10 ani.13 în anumite regiuni ale Spaniei, prevalența ar putea fi chiar mai mare. Pe coasta galiciană, de exemplu, a fost raportată o prevalență de 1 la 33 000, datorită și unui efect fondator.14
ihtioză lamelară și eritrodermie ihtiosiformă Congenitalăcaracteristici clinice
deși s-a crezut inițial că LI și CIE erau entități diferite, au existat raportări ale pacienților cu manifestări clinice intermediare și ambele afecțiuni pot fi cauzate de mutații în aceeași genă.15,16 în plus, pacienții cu aceeași mutație, chiar și în cadrul aceleiași familii, pot dezvolta fenotipuri diferite.12,15
majoritatea pacienților se nasc înveliți într-o membrană collodion care dispare progresiv în primele săptămâni de viață și este înlocuită cu fenotipul definitiv (Fig. 1A). Hipohidroza, intoleranța severă la căldură și distrofia unghiilor sunt frecvent observate atât în LI, cât și în CIE.17-19 pacienți cu LI au de obicei manifestări clinice mai severe decât cei cu CIE. Ei au solzi mari platelike, de multe ori de o culoare închisă, care acoperă întreaga suprafață a corpului. Eritrodermia este fie absentă, fie minimă. Astfel de pacienți au de obicei ectropion și, uneori, eclabiu, hipoplazie a cartilajului articular și nazal, alopecie cicatricială, în special la marginea scalpului și keratoderma palmoplantară (Fig. 1B și C). CIE se caracterizează prin prezența eritrodermiei și a scalării albicioase fine (Fig. 2). Unii pacienți au eritem marcat și scalare generalizată. Cântarele pot fi mari și de culoare închisă, în special pe suprafețele extensoare ale picioarelor. În cazurile mai puțin severe, eritemul este ușor și scalarea este bună.

caracteristicile clinice ale ihtiozei lamelare. O descuamare lamelară maronie. B, hiperkeratoza plantară marcată. C, cicatrizarea alopeciei scalpului.
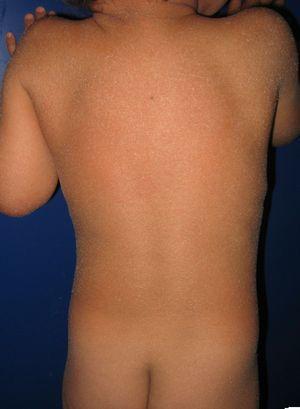
pacient cu eritrodermie ihtiosiformă congenitală și mutații ale genei ALOXE3. Eritem ușor și blană albicioasă generalizatăpoate fi observată descuamarea blănurilor.
Histopatologie
modificările histopatologice nu oferă un diagnostic. În LI, se observă hiperkeratoză ortokeratotică masivă, de obicei cu o extensie de două ori mai mare decât în CIE. Epiderma este acantotică și, ocazional, are un aspect asemănător psoriazisului. Rata de proliferare a celulelor este normală sau ușor ridicată.17-19 pacienți cu CIE au hiperkeratoză mai puțin marcată, cu parakeratoză focală sau extinsă, un strat granular normal sau îngroșat și acantoză mai pronunțată. Cifra de afaceri epidermică este crescută.17-19
ultrastructura
deși o corelație strânsă între constatările moleculare, clinice și ultrastructurale nu a fost găsită până acum, microscopia electronică poate fi totuși utilă pentru excluderea altor forme de ihtioză și pentru ghidarea analizelor genetice în unele cazuri. Au fost descrise patru tipuri de ihtioză congenitală (Tabelul 2).
Clasificarea ultrastructurală a ihtiozelor congenitale.
| Tip | Caracteristică principală | alte caracteristici | mutații | manifestări clinice |
| 1 | absența markerilor ultrastructurali ai ihtiozei tipurile 2, 3 și 4 | picături lipidice sau inele în stratul cornos (cel mai frecvent)granulesvesicular sau lobular granule mici de acoperire cu membrană | TGM1 (33.3%) ALOX12B (2 cazuri) | CIE |
| 2 | clefturi de colesterol în stratul cornos | absența sau subțierea învelișului cornificatmici granule de keratohyalin picături lipidice | TGM1 (89-100%) | LI |
| 3 | structuri membranate laminate în stratul granulos și/sau stratul cornos. | granule anormale de acoperire cu membranăpicături lipidefoci de vacuole juxtanucleare proeminente în stratul granular | NIPAL4 (93%) | CIE (cel mai frecvent)LI |
| 4 | pachete cu membrană Trilamelară care umplu unele celule din stratul granulos și/sau stratul cornos | granule anormale de acoperire cu membrană | FTAP4 | sindromul prematurității Ihtiozei (100%) |
abrevieri: CIE, eritrodermie ihtiosiformă congenitală; LI, ihtioză lamelară.
ihtioza congenitală de tip 1
ihtioza congenitală de tip 1 se caracterizează prin absența markerilor ultrastructurali pentru ihtioza de tip 2, 3 și 4. Prin urmare, diagnosticul se face de obicei numai atunci când celelalte tipuri au fost excluse. Cea mai frecventă constatare este prezența picăturilor sau inelelor lipidice în stratul cornos (Fig. 3A).20 aceste picături lipidice nu sunt o caracteristică constantă sau specifice acestui tip particular,deoarece nu sunt prezente în toate cazurile, 20 și pot fi prezente în alte tipuri de ihtioză.21,22 Clinic, majoritatea pacienților prezintă manifestări de CIE.12,20 o treime dintre pacienți au mutații în gena TGM1.16 Acest tip ultrastructural a fost, de asemenea, identificat în asociere cu mutații ale genei ALOX12B.23,24

imagini cu microscop electronic. A, ihtioza congenitală de tip 1, care prezintă picături lipidice în stratul cornos și absența markerilor ultrastructurali ai celorlalte tipuri de ihtioză. B, ihtioza congenitală de tip 2, caracterizată prin prezența crăpăturilor de colesterol (săgeată) în corneocite.
ihtioza congenitală de tip 2
ihtioza congenitală de tip 2 se caracterizează prin fisuri de colesterol în stratul cornos (Fig. 3B).21 astfel de fisuri sunt o constatare constantă în acest tip de ihtioză și pot fi detectate în diferite biopsii la același pacient; tratamentul cu retinoizi orali nu are niciun impact asupra acestor fisuri.12,25 agregate dense de electroni au fost, de asemenea, observate pe corneocite la unii pacienți cu activitate deficitară a Tgazei 1.26-28 Clinic, majoritatea pacienților prezintă manifestări severe de CIE.12 acest tip ultrastructural este puternic asociat cu mutații ale genei TGM1.12,16
ihtioza congenitală de tip 3
ihtioza congenitală de tip 3 se caracterizează prin structuri membranoase lamelare în stratul granulos și/sau stratul cornos. Aceste structuri sunt aranjate în benzi în jurul unui spațiu gol aproape de nucleu.22,29-31 manifestările clinice la acest tip sunt diferite de celelalte; debutul ihtiozei este variabil, descuamarea și eritemul pot fi neuniforme sau generalizate, iar flexurile în special sunt afectate. Mutațiile genei NIPAL4 sunt responsabile pentru 93% din ihtiozele de tip 3.32
ihtioza congenitală de tip 4
caracteristic, în ihtioza congenitală de tip 4, unele celule din stratul granulos și stratul cornos sunt umplute cu pachete de membrană trilamelară.33 aceste constatări sunt patognomice pentru sindromul prematurității ihtiozei, o afecțiune considerată în prezent ca o formă sindromică de ihtioză.34,35
studii moleculare
în termeni genetici, ARCIs sunt foarte eterogene. Gena TGM1 este asociată cu majoritatea cazurilor, dar au fost raportate mutații în alte 5 gene (ALOX12B, ALOXE3, NIPAL4, CYP4F22 și ABCA12). Fischer și colab.36 au studiat 520 de familii cu ARCI și au identificat mutații în cel puțin 1 dintre aceste gene în 78% din cazuri (TGM1 în 32%, NIPAL4 în 16%, ALOX12B în 12%, CYP4F22 în 8%, ALOXE3 în 5% și ABCA12 în 5%). Într-un alt studiu efectuat pe 250 de pacienți cu ARCI de origini diferite, 38% au avut mutații TGM1, 6,8% au avut mutații ALOXE3 și 6,8% au avut mutații ALOX12B.37 În Galicia, am identificat mutații în genele TGM1, ALOX12B, ALOXE3, NIPAL4 și CYP4F22 în 75% din familiile studiate, dar distribuția mutațiilor a fost diferită.14 gena TGM1 a fost mutată în 68.7% din cazuri în timp ce gena ALOXE3 a fost mutantă la doar 1 pacient. Nu am detectat mutații în niciuna dintre celelalte 3 gene studiate.
TGM1
gena TGM1 este localizată pe cromozomul 14q11.2 și are 15 exoni (GenBank NM-000359.2). Codifică enzima TGase 1, Care este una dintre cele 3 enzime TGase găsite în epidermă.38 această enzimă participă la formarea învelișului cornificat prin catalizarea reticulării dependente de calciu a mai multor proteine, cum ar fi involucrina, loricrina și proteinele bogate în prolină.39,40 de asemenea, catalizează legarea ??- hidroxiceramide în stratul exterior al învelișului cornificat cu proteine în stratul interior.41,42 la pacienții cu mutații TGM1, învelișul cornificat lipsește și activitatea Tgazei 1 este redusă sau inexistentă.43-47
din 1995, când această genă a fost identificată ca fiind responsabilă pentru unele cazuri de ARCI,48-50 mai mult de 110 mutații au fost raportate la pacienții de origini diferite. Mutațiile în TGM1 sunt cea mai frecventă cauză a ARCI.36,37 această mutație a fost găsită în 55% din cazuri în Statele Unite și în 84% din cazuri în Norvegia.12,51 cea mai frecventă mutație este c.877-2a>G, care a fost găsit în 34% din alelele mutante raportate până în prezent.52 frecvența ridicată a acestei mutații în țări precum Statele Unite și Norvegia se datorează unui efect fondator.12,53 a doua mutație cea mai frecventă este P.Arg142His. Acest lucru și mutații similare au fost raportate în țări precum Egipt, Germania, Finlanda și Statele Unite, 15,49-51,54-56 și s-ar părea că acestea sunt mutații hotspot.57 mutația P. Arg307Trp este frecventă la populația japoneză.5 În Galicia, p. Arg760X, c. 1223_1227delACACA și c.984 + 1g >a mutații în TGM1 au fost identificate în 81,82% din familiile cu mutații în această genă, sugerând un efect fondator.14 confirmarea acestei ipoteze a fost obținută prin studiul haplotipului (lucrare încă nepublicată).
mutațiile TGM1 sunt responsabile pentru majoritatea cazurilor de LI15,27,44,46,56,58-63 și pentru un procent mic de cazuri de CIE.43,47,64,65 astfel de mutații pot da naștere și altor forme de ARCI, cum ar fi SHCB, acral SHCB și ihtioza costumului de baie.
multe studii au încercat să demonstreze asocierile genotip-fenotip între mutațiile tgm1 și constatările ultrastructurale sau clinice, dar până în prezent nu a fost observată nicio corelație semnificativă.15,16,53 în general, pacienții cu mutații ale genei TGM1 sunt mai grav afectați decât cei fără astfel de mutații. Într-un studiu efectuat pe 83 de pacienți cu ARCI din Suedia și Estonia, prezența ectropionului și a colodionului baby a fost asociată cu mutații TGM1, în timp ce o rată mai mare de eritem a fost observată la pacienții fără mutații în această genă.66 un alt studiu a arătat că tipul de scalare este principala diferență între purtătorii și cei care nu poartă mutații TGM1, constatând că toți pacienții cu mutații în această genă au avut scalare lamelară, în timp ce 80% dintre cei fără mutații TGM1 au avut scalare fină.14 în plus, s-a observat că mutațiile trunchiate sunt mai frecvent asociate cu hipohidroza și tulburările de transpirație decât mutațiile missense.51 în populația nord-americană, un model bazat pe prezența anumitor caracteristici clinice prezice că pacienții care se nasc ca copii collodion și au tulburări oculare și/sau alopecie sunt de 4 ori mai predispuși să aibă mutații TGM1.51
aloxe3 și ALOX12B
genele ALOXE3 și ALOX12B sunt localizate pe cromozomul 17p13.1.67 au o structură similară cu 15 exoni care codifică lox-urile epidermice eLOX-3 și 12R-LOX.68,69 faptul că acestea sunt exprimate predominant în straturile suprabasale ale epidermei susține rolul lor în fazele avansate de diferențiere epidermică, cu participarea la prelucrarea corpurilor lamelare.24,70 aceste enzime acționează pe treptele adiacente ale căii hepoxilinei (Fig. 4). 12R-LOX transformă acidul arahidonic în acid 12R-hidroxieicosatetraenoic, în timp ce eLOX-3 transformă acest produs într-un izomer epoxialcool69,71 din familia hepoxilin A3.72 produsul hepoxilin este instabil și este hidrolizat în celule la un derivat trihidroxi specific (trioxilin). Deși rolul exact al produselor căii hepoxilinei nu este cunoscut, s-a speculat că acestea pot participa la formarea lipidelor intercelulare ale stratului cornos sau pot acționa ca semnale pentru inducerea diferențierii keratinocitelor.
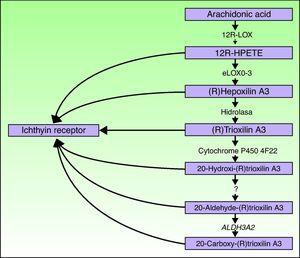
diagrama schematică a căii hepoxilinei, care arată participarea genelor ALOXE3, ALOX12B, NIPAL4 și CYP4F22. Mutațiile acestor gene sunt responsabile pentru unele tipuri de ARCI. HPETE indică acidul hidroperoxieicosatetraenoic.
genele ALOX12B și ALOXE3 au fost identificate pentru prima dată în 2002.73,74 de atunci, au fost raportate mai mult de 30 de mutații în geneza ALOX12B23,24,37,75-77 și aproximativ 10 în geneza ALOXE337,74,75. Aceste mutații sunt responsabile pentru 14% până la 17% din ARCIs36,37 și 72.2% din SHCBs.23,78,79 relația cauzală dintre aceste mutații și fenotip a fost confirmată prin demonstrarea faptului că activitatea catalitică a LOXULUI epidermal a fost complet eliminată la pacienții cu aceste mutații75,80 și prin utilizarea modelelor animale care au reprodus fenotipul ihtiosiform observat la om.81-83 ambele gene sunt responsabile pentru un procent similar de cazuri ARCI. Cu toate acestea, gama de mutații diferite în gena ALOXE3 este limitată, datorită predominanței a 2 mutații, P.Arg234X și P.Pro630Leu, care par să corespundă punctelor fierbinți.37,74,75
pacienții cu mutații ale genelor ALOXE3 și ALOX12B prezintă de obicei un fenotip CIE.74,75,77 severitatea scalării este ușoară sau moderată, iar solzii au o culoare albicioasă sau maro deschis. Eritemul poate fi, de asemenea, prezent. Până la 76% dintre pacienți se nasc ca bebeluși collodion și 88% au tulburări de transpirație.37 de pacienți cu mutații în gena ALOX12B prezintă o descuamare mai limitată, albicioasă, comparativ cu purtătorii de mutații din gena ALOXE3. În aceste cazuri, solzii sunt maronii și aderenți. Prezența eritemului, hiperkeratozei palmoplantare și accentuarea pliurilor palmoplantare sunt, de asemenea, asociate cu mutații ALOX12B.37
Ihtiină/NIPAL4
gena NIPAL4, cunoscută și sub numele de gena ihtiinei, este localizată pe cromozomul 5q33. Are 6 exoni care codifică o proteină cu mai multe domenii transmembranare cu funcție necunoscută.84 s-a emis ipoteza că produsul proteic participă la aceeași cale metabolică ca LOX și poate acționa ca un receptor pentru trioxilinele A3 și B3 sau pentru alți metaboliți ai căii metabolice a hepoxilinei.84 ar fi astfel implicat în formarea corpurilor lamelare sau în transportul acestora către spațiul extracelular.32 În sprijinul acestui lucru sunt 2 observații. În primul rând, în 93% din cazuri, mutațiile acestei gene sunt asociate cu un model ultrastructural de ihtioză congenitală de tip 3, caracterizată prin anomalii ale corpurilor lamelare și prezența membranelor perinucleare alungite în stratul granulosum.32 În al doilea rând, NIPAL4 este exprimat în esență în stratul granulos al epidermei, unde sunt prezente corpurile lamelare.85
de la descoperirea genei NIPAL4 în 2004,84 au fost raportate doar 9 mutații la pacienții din țările mediteraneene (Algeria, Turcia și Siria), 84 de țări scandinave,32 Pakistan,85 Insulele Feroe,32 și America de Sud.84
spectrul clinic al pacienților cu mutații în această genă este larg, chiar și în rândul membrilor aceleiași familii. Între 3,7% 32 și 60% 84 se nasc ca copii collodion. Când membrana colodionului dispare, majoritatea pacienților dezvoltă manifestările CIE, cu solzi albicioși fini pe o bază eritematoasă pe față și trunchi și solzi mai mari, maronii, pe gât, fese și picioare.84 xeroză marcată, plăci hiperkeratotice reticulare maronii generalizate care apar accentuate în pliurile pielii și pot fi prezente dischromii faciale.32,85 în plus, keratoderma palmoplantară este o constatare frecventă, împreună cu contracturile ocazionale ale degetelor și unghiile curbate ale degetelor. Unele studii au raportat rezultate mai tipice pentru LI.32,85 prezența semnelor și simptomelor dermatitei atopice a fost raportată la unii pacienți, deși mutațiile genei FLG nu au fost detectate în niciunul dintre aceste cazuri.85
CYP4F22
gena FLJ39501 sau cyp4f22 este localizată pe cromozomul 19p13.12.86 are 12 exonuri87 și codifică un citocrom P450, familia 4, subfamilia F, polipeptida 2, omolog al leucotrienei B4 – XV-hidroxilazei (CYP4F2). Reacția catalizată de produsul flj39501 la nivelul pielii și substraturile acestei reacții poate fi dedusă prin analogie cu omologii săi cunoscuți CYP4F2 și CYP4F3.88 s-a emis ipoteza că CYP4F2 și CYP4F3 participă la calea hepoxilinei prin catalizarea conversiei trioxilinei A3 în 20-hidroxi-(R)trioxilină A387 și că produsul final al acestei căi, 20-carboxi-trioxilina A3, poate avea un efect biologic cheie de reglare la nivelul pielii.89
până în prezent, doar 8 mutații ale acestei gene au fost raportate în 12 familii consanguine din țările Mediteraneene87 și în 1 familie de origine israeliană.62
în familiile raportate de către Leftecicvre și colab., 87 majoritatea pacienților au avut un fenotip CIE la naștere și acest lucru a progresat ulterior la LI. pacienții s-au născut de obicei cu eritrodermie marcată, deși fără membrană de colodion. Pe măsură ce au îmbătrânit, au dezvoltat scalarea generalizată alb-cenușie, care a fost mai marcată în regiunea periumbilicală, pe fese și pe partea inferioară a corpului. Hiperlinearitatea palmelor și tălpilor și descuamarea pe scalp, în momente de tip pityriasiform, au fost frecvente.87 într-o altă familie, cei 3 membri afectați s-au născut ca bebeluși de coliziune și au dezvoltat eritrodermie intensă, descuamare generalizată și keratodermă palmoplantară.62
ABCA12
în 2003, gena ABCA12 a fost raportată a fi responsabilă pentru unele cazuri de LI și a fost mapată la cromozomul 2q34.4 ulterior s-a confirmat că mutațiile din această genă au fost, de asemenea, responsabile pentru HI.2, 3ABCA12 codifică 53 de exoni și aparține unei familii de transportori ABC, care leagă adenozin trifosfatul, facilitând în același timp transportul mai multor molecule prin membrana celulară.90 Membrii subfamiliei ABCA sunt implicați în transportul lipidelor.91 funcția ABCA12 deficitară provoacă tulburări de transport lipidic în corpurile lamelare și astfel duce la o scădere a nivelului lipidelor intercelulare în stratul cornos.3studiile ultrastructurale au arătat că ABCA12 este localizat în corpuri lamelare asociate cu glicozilceramide.91abca12 mutațiile au fost asociate cu tulburări în distribuția și transportul glicozilceramidelor și cu niveluri scăzute de hidroxiceramide, una dintre componentele principale ale barierei lipidice din spațiile intercelulare.3,6,92,93 hiperkeratoza masivă care apare la acești pacienți ar putea fi un răspuns compensatoriu la o barieră lipidică deficitară.94 s-ar putea datora și lipsei descuamării corneocitelor,93 care ar putea fi cauzată de defecte în transportul anumitor proteaze, cum ar fi callicreina 5 și catepsina D, care rezultă din tulburări în corpurile lamelare.95 de modele Murine și studii in vitro sugerează că mutațiile ABCA12 au, de asemenea, un efect asupra diferențierii epidermice.95-97
până în prezent, mai mult de 50 de mutații au fost raportate în gena ABCA12 la pacienții cu ARCI din Africa, Europa, Pakistan și Japonia. Cele mai frecvente mutații sunt P.Val244SerfsTer28,2,98,99 identificate în populațiile pakistaneze și indiene și p.Asn1380Ser, 4 identificate în familiile africane. În ambele cazuri, acestea pot fi mutații fondatoare.
amploarea mutațiilor ABCA12 este legată de fenotip, cu mutații asociate cu pierderea completă a funcției care duce la fenotipul HI.2,3,98 – 102 în schimb, în LI și CIE, majoritatea mutațiilor sunt missense și au un efect mai puțin sever asupra funcției proteinelor.4-6, 103 mutațiile care stau la baza fenotipului LI par a fi concentrate în prima regiune a casetei de legare a adenozin trifosfatului.4 din punct de vedere clinic, pacienții cu CIE și mutații ale genei ABCA12 au scale de dimensiuni medii care sunt ceva mai mari decât cele observate de obicei la pacienții cu acest fenotip.
ihtioza Arlequin
HI sau fătul Arlequin este o formă severă și de obicei fatală de ihtioză. Copiii sunt de obicei prematuri cu plăci hiperkeratotice strălucitoare extinse, separate prin fisuri adânci, care acoperă întregul integument și formează modele geometrice care amintesc de îmbrăcămintea purtată de arlechini, dând astfel condiției numele său. Etanșeitatea pielii duce la o eversiune marcată a pleoapelor și buzelor, la dezvoltarea rudimentară a cartilajului articular și nazal și, ocazional, la microcefalie. Copiii au rareori gene sau sprâncene, deși părul de pe scalp poate fi conservat. Mâinile și picioarele sunt umflate și edematoase și adesea acoperite de un strat asemănător mănușii. Pot avea contracturi cu degetele.
pentru astfel de pacienți, riscul de a muri în perioada neonatală este foarte mare.104 ventilația pulmonară este compromisă; pierderea de apă transepidermică duce la deshidratare, dezechilibru hidroelectric și instabilitate termică; iar riscul de infecții este crescut. Etanșeitatea facială și eclabiul împiedică suptul și, prin urmare, hrănirea, cu agravarea corespunzătoare a deshidratării. Nou-născuții cu această afecțiune rareori au trăit mai mult câteva săptămâni. Cu toate acestea, în ultimii ani, șansele de supraviețuire pe termen lung au crescut în special, în principal datorită administrării retinoizilor sistemici și progresului în îngrijirea intensivă neonatală.105 într-un studiu recent, 83% dintre pacienții tratați cu retinoizi orali au supraviețuit, comparativ cu 24% dintre pacienții netratați. Majoritatea deceselor au avut loc în primele 3 zile de viață, dar tratamentul nu a fost început decât după aceasta la mulți dintre supraviețuitori.104 acest lucru ar sugera că multe dintre aceste decese timpurii ar fi avut loc indiferent de tratamentul retinoid.
copiii care supraviețuiesc perioadei neonatale dezvoltă în general CIE severă.106 natura și localizarea mutațiilor genei ABCA12 și amploarea pierderii funcției transportorului pot determina prognosticul.3.92.107 pacienți care conservă un anumit grad de activitate proteică, deși minim, pot avea șanse mai mari de supraviețuire. Purtătorii de mutații homozigote au o rată de mortalitate mai mare.104
principala caracteristică histologică a HI este prezența unui strat cornos ortokeratotic extrem de gros și compact. Foliculii de păr și conductele de transpirație au dopuri hiperkeratotice proeminente107, 108 și au corpuri lamelare anormale sau absente, incluziuni lipidice sau resturi de organite sau nuclee în corneocite și absența lipidelor intercelulare în studiul ultrastructural.108.109 foliculii de păr prezintă o concentrație marcată de material keratotic, care este o caracteristică diagnostică a HI utilizată pentru diagnosticul prenatal.
până în prezent, rata de detectare a mutațiilor în gena ABCA12 la pacienții cu HI este aproape de 100%, deci aceasta pare a fi o condiție omogenă genetic.
copilul Collodion și copilul Collodion auto-vindecător
copiii Collodion se nasc de obicei prematur, iar morbiditatea și mortalitatea perinatală sunt crescute. La naștere, nou-născutul este acoperit de o membrană transparentă învățată strălucitoare care amintește de ambalajul celofan (Fig. 5). Bebelușii au ectropion, eclabiu și hipoplazie a cartilajului nazal și articular. Sugerea și ventilația pulmonară pot fi împiedicate110 și pierderea transepidermică de apă și riscul de infecții sunt crescute.110,111

copil Collodion care ulterior a progresat la un fenotip de ihtioză lamelară.
Collodion baby este prezentarea obișnuită pentru HI și CIE. Autozomal dominant LI, 112, 113 SJ sindromul Xvgren-Larsson, 110 trichothyodystrophy,114 boala Gaucher juvenilă, 110 boala de stocare a lipidelor neutre, sindromul Conradi-h, sindromul Hays-Wells și displazia ectodermică115 pot, de asemenea, să apară ocazional ca bebeluș collodion. Membrana dispare spontan la 10% până la 24% dintre nou-născuți, pentru a da loc pielii complet normale.110.116 în trecut, aceste cazuri au fost descrise ca LI al nou-născutului,117, dar nu sunt denumite SHCB.118 unii autori au sugerat termenul de ihtioză colodială auto-amelioratoare, deoarece mulți dintre acești pacienți, atunci când sunt reexaminați mai târziu în copilărie sau ca adulți, au un grad variabil de anhidroză și intoleranță la căldură și semne ușoare de ihtioză, cum ar fi xeroza și descuamarea fină, în special în axile și gât.78
nici microscopia optică, nici investigațiile ultrastructurale ale copilului collodion nu sunt specifice. Prin urmare, este de preferat să întârzieți biopsia cutanată până la dezvoltarea fenotipului definitiv.
mutații ale genelor TGM1,7,119aloxe3,78 și ALOX12B23,78,79 au fost identificate la pacienții cu SHCB. Mutațiile ALOX12B sunt cele mai frecvente. Într-o serie de 15 pacienți scandinavi cu SHCB, 67% au avut mutații în gena ALOX12B, 25% în gena ALOXE3 și 8,3% în gena TGM1.78 de mutații nu au fost găsite la unii pacienți și, prin urmare, este posibil ca și alte gene să fie implicate. S-a speculat că aceste mutații reduc activitatea enzimatică în uter, dar nu după naștere.7 în uter, unde presiunea hidrostatică este ridicată, chelarea prin apă transformă enzima mutantă într-o conformație inactivă. După naștere, când presiunea scade, enzima revine la forma sa activă și activitatea sa crește suficient pentru a menține un fenotip normal sau minim afectat.7
Acral auto-vindecare collodion Baby
deși collodion baby afectează întregul corp, au fost raportate cazuri limitate la regiunile acrale. În 1952, Finlay și colab.120 a raportat un caz de membrană collodion care a afectat doar mâinile și picioarele și care a urmat un curs de auto-vindecare. Recent, un nou caz de shcb acral a fost raportat în asociere cu mutații ale genei TGM1.8 nu se știe de ce aceste leziuni sunt limitate la regiunile acrale, deși factorii asociați cu reglarea dependentă de situs a activității enzimatice pot fi în funcțiune.8
ihtioza costumului de baie
ihtioza costumului de baie a fost raportată pentru prima dată ca variantă Arci independentă în 2005, deși au fost raportate anterior cazuri de ihtioză cu o distribuție specifică.121-123 a fost detectat în principal la pacienții de origine sud-africană,9 deși a fost raportat și la persoane din Europa și țările mediteraneene.124 la naștere, pacienții au o membrană colodion generalizată care apoi se varsă pentru a părăsi distribuția caracteristică a scalării. Trunchiul, Regiunea proximală a brațelor, inclusiv axilele, gâtul și scalpul sunt în general afectate, în timp ce partea centrală a feței, a membrelor și a regiunii suprarenale sunt de obicei scutite.9 cântarele sunt mari, lamelare și de culoare închisă. Descuamarea mai fină poate apărea în fosa popliteală și antecubitală.124.125 palmele mâinilor și tălpilor picioarelor au hiperkeratoză difuză ușoară, în timp ce spatele mâinilor și picioarelor nu prezintă nicio implicare.
studiul histopatologic al pielii afectate prezintă hiperkeratoză marcată fără parakeratoză, straturi granulare normale, acantoză ușoară sau moderată și un infiltrat limfocitar ușor în dermul superior.9 observațiile cu microscopie electronică sunt în concordanță cu ihtioza congenitală de tip 2 în majoritatea cazurilor. Pielea neimplicată nu prezintă Constatări anormale.124.125 în pielea sănătoasă, activitatea TGase 1 este ușor redusă și, de obicei, localizată în zonele pericelulare. În pielea implicată, activitatea enzimatică este reziduală și localizată anormal în citoplasmă.124
mutații au fost detectate în gena TGM1 la toți pacienții cu ihtioză în costum de baie studiată până în prezent.119.124-126 cea mai frecventă mutație este P.Arg315Leu, care a fost identificată la majoritatea pacienților din Africa de Sud și ar putea fi o mutație fondatoare. Oji și colab.124 a sugerat că temperatura pielii ar putea juca un rol în dezvoltarea acestor manifestări. Folosind termografia digitală, autorii au arătat o corelație puternică între temperatura corpului și descuamare, cele mai fierbinți zone ale corpului fiind cele mai afectate. Aufenvenne și colab.127 a arătat o scădere a temperaturii optime pentru activitatea TGase 1 la pacienții cu ihtioză a costumului de baie. Această scădere nu a fost observată la controalele sănătoase sau la pacienții cu LI generalizată. această scădere a temperaturii ar explica fenotipul acestor pacienți. Temperatura optimă este de 37% C pentru enzima normală, dar de 31% C pentru enzima mutantă.
tratament
scopul principal al tratamentului în ihtioză este eliminarea scalării și reducerea xerozei fără a provoca iritații excesive (Tabelul 3). Înainte de a decide cu privire la tratament, trebuie luate în considerare aspecte precum vârsta și sexul pacientului, tipul și severitatea bolii, precum și amploarea și locul leziunilor.128
strategia terapeutică în Ihtiozele congenitale autosomale recesive.
| strategia terapeutică pentru ihtiozele congenitale autozomale recesive | |
| scăldat și eliminarea mecanică a cântarelor | scăldat cu bicarbonat de sodiu sau amidon de grâu, amidon de porumb sau amidon de orez; îndepărtarea mecanică a cântarelor (de 1 sau 2 ori pe zi) |
| tratament topic (secvențial) | hidratante care conțin Ureiceratinolitice cu propilen glicolceratinolitice combinate (propilen glicol, acizi hidroxi-uri sau uree) Keratinolitice combinate cu acid salicilicretinoizii topicila nou-născuți și copii mici, aplicați un vehicul fără ingrediente active. Evitați ureea, acidul salicilic și acidul lactic din cauza riscului de absorbție sistemică |
| tratament Oral | retinoide orale (acitretină sau izotretinoină) |
| alte măsuri | urmărirea ectropionului de către oftalmologcurățarea regulată a urechii externe de către specialistul ureche-gât-nasfizioterapie pentru prevenirea contracțiilor.Evitarea activităților intense într-o temperatură ambientală ridicată |
scăldatul și eliminarea mecanică a cântarelor
scăldatul zilnic este recomandat pacienților cu ARCI pentru a elimina mecanic solzii și urmele de hidratant. Acest lucru este mai ușor dacă pacientul este scufundat în apă timp de 15 până la 30 de animinute. Unii autori recomandă adăugarea bicarbonatului de sodiu în baie pentru a denaturaliza keratinele și a face apa alcalină, facilitând astfel eliminarea solzilor.129 alte produse care pot fi adăugate includ amidon de grâu, amidon de porumb sau amidon de orez. Uleiurile pentru scăldat nu sunt adecvate, deoarece pot duce la ocluzie, cu risc ulterior de proliferare bacteriană și agravarea termoreglării.
tratament topic
Hidratantele și agenții keratolitici topici sunt de obicei prima opțiune terapeutică. Îmbunătățește funcția de barieră a pielii și facilitează descuamarea. Pot apărea reacții adverse locale ușoare, cum ar fi prurit tranzitoriu, iritație sau senzație de înțepătură.
clorura de sodiu, ureea, acetat de vitamina E, glicerol și vaselină pot fi utilizate ca hidratante și lubrifianți. La pacienții cu scalare groasă și hiperkeratoză marcată, se pot adăuga 1 sau mai mulți agenți keratolitici, cum ar fi acizi hidroxi-hidroxi (acid lactic și glicolic),130 acid salicilic, N-acetilcisteină,131-133 uree (>5%),134 și propilen glicol. Se utilizează, de asemenea, modulatori de diferențiere a keratinocitelor. Acestea includ retinoizi topici (tretinoin, adapalen, tazaroten),135,136 calcipotriol,137 și dexpanthenol.Retinoizii topici provoacă adesea iritații și fisuri mici, foarte dureroase.137 în plus, există un risc de absorbție și teratogenitate la femeile fertile dacă acestea sunt utilizate prea mult.138 pentru a spori eficacitatea keratoliticelor și hidratantelor, pansamentul ocluziv poate fi aplicat în zone specifice refractare la tratament.139 un efect aditiv sau sinergic poate fi obținut și prin combinarea a 2 sau mai mulți agenți keratolitici sau hidratanți.140-142 tratamentul trebuie optimizat pentru fiecare individ, având în vedere natura foarte variabilă a afecțiunii și sensibilitatea pielii și diferențele de răspuns la fiecare tratament. Procesul de optimizare poate fi ajutat prin tratarea unei părți a corpului diferit față de cealaltă pentru a permite comparații. Nou-născuții și copiii mici trebuie tratați cu un vehicul fără substanțe active, deoarece pielea este foarte fină și sensibilă și majoritatea keratoliticelor nu sunt tolerate. În plus, riscul de absorbție percutanată a produselor topice precum ureea, acidul salicilic și acidul lactic este mai mare.143-145
tratamentul sistemic
retinoizii orali au efecte keratolitice care ajută la eliminarea scalelor și la prevenirea hiperkeratozei excesive. Atât izotretinoina, cât și retinoizii aromatici (acitretină și etretinat) s-au dovedit eficiente în tratamentul ARCIs.128.146.147 Acitretin la o doză de 0,5 până la 1 mg/kg/zi este cel mai utilizat medicament, în special la pacienții cu LI.148 pacienții cu CIE pot avea un răspuns mai complet și la doze mai mici.
principalele efecte adverse sunt tulburările mucocutanate, teratogenitatea, tulburările musculo-scheletice și profilul lipidic anormal și creșterea transaminazelor.149-152 în ceea ce privește teratogenitatea, în cazul etretinatului și acitretinei, medicamentele trebuie evitate în timpul sarcinii, iar pacienții trebuie să evite să rămână gravide timp de 3 ani după întreruperea tratamentului.151 isotretinoina are un timp de înjumătățire mai scurt și este complet eliminată din organism după 1 lună și astfel poate fi opțiunea preferată la femeile care doresc să rămână gravide.128
monitorizarea tratamentului trebuie să includă un studiu de laborator cu un test al funcției hepatice și al profilului lipidic înainte de începerea tratamentului, apoi la 1 lună și la fiecare 3 luni după începerea tratamentului. La femeile fertile, trebuie efectuat un test de sarcină în cele 2 săptămâni înainte de începerea tratamentului și trebuie utilizată o măsură contraceptivă eficientă de la 4 săptămâni înainte de tratament până la 3 ani după aceea (în cazul acitretinei). Când este necesar un tratament prelungit cu retinoizi, trebuie monitorizată creșterea și dezvoltarea osoasă. Unii autori sugerează efectuarea unui studiu osos înainte de tratament, urmat de o examinare anuală.151 ghidurile recente nu recomandă efectuarea radiografiei de rutină din cauza posibilelor efecte dăunătoare.152 în schimb, se recomandă studii radiografice selective la pacienții cu dureri osoase atipice.152
o alternativă la tratamentul retinoid sistemic este utilizarea de medicamente cunoscute sub numele de agenți de blocare a metabolismului acidului retinoic, care cresc nivelurile endogene ale acidului retinoic. Un astfel de medicament este liarozol, căruia i s-a acordat statutul de orfan pentru tratamentul LI, CIE și HI de către Agenția Europeană pentru medicamente și Administrația SUA pentru alimente și medicamente.153-155 acest medicament s-a dovedit a fi mai eficient decât acitretina în studiile clinice și este, de asemenea, mai bine tolerat și are un profil farmacocinetic mai bun.154
alte îngrijiri medicale
la pacienții cu ectropion, aplicarea lacrimilor artificiale și a lubrifianților pentru ochi și hidratarea pielii feței și a obrajilor în special poate reduce retragerea palpebrală. Corecția chirurgicală este o opțiune valabilă în cazurile severe, dar aceasta trebuie repetată de obicei câțiva ani mai târziu. Hidroterapia poate fi benefică.156 de pacienți trebuie sfătuiți să evite activitatea fizică intensă atunci când temperatura ambiantă este ridicată, având în vedere că hipohidroza implică riscul de accident vascular cerebral și convulsii. Retinoizii orali pot îmbunătăți termoreglarea.157 fizioterapia este importantă pentru prevenirea contracției de flexie, în special în cazul HI. Curățarea regulată a canalului auditiv extern de către un specialist în ureche-gât-nas poate preveni acumularea cântarelor și astfel poate preveni pierderea auzului.
consiliere genetică și diagnostic Prenatal
când un pacient este diagnosticat cu ihtioză, trebuie să i se ofere consiliere genetică adecvată în care să fie explicate natura tulburării, modul de transmitere și riscul manifestărilor viitoare în familie. Diagnosticul Prenatal poate indica dacă fătul este afectat și, dacă este cazul, poate fi oferită pregătirea psihologică a familiei și pot fi anticipate probleme în timpul sarcinii și nașterii. Părinților li se poate oferi opțiunea unui avort dacă nu este disponibil un tratament. În plus, dacă terapia genică pentru aceste afecțiuni va deveni disponibilă în viitor, diagnosticul prenatal ar permite aplicarea acestei terapii cât mai curând posibil.
pentru mai mult de 20 de ani, diagnosticul prenatal a fost efectuat prin prelevarea unei probe de biopsie a pielii fetale și studierea acesteia prin microscopie optică, microscopie electronică sau imunohistochimie.158.159 această procedură invazivă a putut fi efectuată numai în fazele târzii ale sarcinii, între săptămânile 15 și 23 de gestație și a fost asociată cu un risc de 1% până la 3% de a pierde fătul.160.161 identificarea mecanismelor moleculare ale afecțiunilor ereditare ale pielii a permis un diagnostic mult mai devreme bazat pe tehnici genetice.102.162-164 ADN Fetal se obține prin amniocenteză efectuată între săptămânile 15 și 20 sau prin prelevarea de villus corionic între săptămânile 10 și 12. Riscul pierderii fetale cu aceste tehnici este mai mic decât între 0,5% și 1%.165 alte metode neinvazive în dezvoltare sunt analiza ADN-ului celular fetal și a ADN-ului fetal liber în circulația maternă166, precum și utilizarea ultrasunetelor 3-dimensionale.167.168
diagnosticul genetic de preimplantare ar putea fi posibil și în tehnicile de fertilizare in vitro, astfel încât numai ouăle fertilizate fără mutație sunt implantate în uter, evitând astfel necesitatea avortului în majoritatea cazurilor.169
strategii viitoare pentru tratamentul Genetic al Ihtiozei
deși s-au înregistrat progrese importante în diagnosticul genetic al ihtiozei, se urmăresc și noi strategii pentru aceste boli.170 pielea este organul cel mai accesibil pentru terapiile de transfer de gene, astfel încât astfel de tehnici sunt minim invazive.171 cu toate acestea, pielea are, de asemenea, caracteristici imunologice unice care nu favorizează expresia pe termen lung a unui produs transgenic.172 în LI, un proces de transfer de gene ex vivo a reușit să restabilească expresia normală TGM1 și să corecteze fenotipul pielii transplantate pe spatele șoarecilor imunosupresați.173.174 recent, fenotipul keratinocitelor cultivate de la pacienții cu HI datorită mutațiilor genei ABCA12 a fost, de asemenea, recuperat.3
conflicte de interese
autorii declară că nu au conflicte de interese.