frontiere în neurologie
Introducere
Neuroacantocitoza este un termen superordonat pentru un grup de sindroame rare care se caracterizează prin simptome neurologice în combinație cu celule roșii din sânge deformate (acantocite). Acest raport se concentrează asupra Coreei-acantocitoză (ChAc) , care este o boală orfană cu aproximativ 1.000 de persoane afectate la nivel mondial cauzate de mutații autozomal-recesive în gena VPS13A (vacuolar protein sorting 13 homolog a) pe cromozomul 9q (1). Boala are un curs progresiv, cauzele mortalității crescute pot fi declinate funcțiile motorii corelate cu condițiile de risc, cum ar fi disfagia, dar au fost descrise și decese bruște neașteptate (2, 3). Până în prezent, nu există un tratament cauzal.
coexistența suspectă a acantocitozei și a tulburărilor de mișcare a fost raportată pentru prima dată în anii 1970 de Levine și Critchley (4, 5) și de atunci a fost acceptată ca trăsătură proeminentă a bolii. Cu toate acestea, cu raportul nostru de caz descriem un fenotip clinic rar de ChAc lipsit de tulburări de mișcare evidente, sugerând o varietate mai largă de prezentări clinice. Instrumentele de diagnosticare trebuie să facă față acestor provocări pentru a stabili diagnosticul corect, sugerăm aici neuroimagistica moleculară ca metodă cheie.
raport de caz
pacientul cu indice masculin a prezentat pentru prima dată la vârsta de 25 de ani două crize tonice clonice bilaterale neprovocate. Nu a existat nici un istoric medical. Scanarea RMN a creierului și EEG la acea vârstă au fost normale și nu a fost inițiat niciun tratament din cauza reticenței pacientului și a evenimentelor rare. Cu toate acestea, pacientul a avut convulsii în curs de desfășurare prezentându-se acum ca epilepsie parțială. El a descris reapariția sentimentelor de amețeală bruscă care au fost diagnosticate ca o aură vertiginoasă. Mai mult, el a avut convulsii discognitive în care a arătat fie (a) lipsa de reacție și recitarea rugăciunilor turcești, (b) automatisme orale, fie (c) jucându—se cu mâinile și rostind sunete-toate evoluând parțial în convulsii tonice clonice. Video-EEG a arătat acum complexe locale de unde spike atât în lobul temporal drept, cât și în cel stâng. În conformitate cu Semiologia convulsiilor, s-a pus diagnosticul de epilepsie bilaterală a lobului temporal și s-a început medicația cu levetiracetam.
la vârsta de 31 de ani convulsiile nu au fost suprimate în totalitate, ca parte a diagnosticării ulterioare a epilepsiei lobului temporal, pacientul a suferit un FDG-PET (Figura 1) care a prezentat un hipometabolism meziotemporal bilateral, în conformitate cu originea convulsiilor meziotemporale. Cu toate acestea, a existat și un hipometabolism striatal marcat, extrem de neobișnuit, care a ridicat suspiciunea unei tulburări de mișcare neurodegenerativă. În consecință, a fost inițiată o evaluare extensivă de monitorizare (a se vedea Figura 2). S-a efectuat un FP-cit-SPECT care a determinat o pierdere moderată bilaterală a disponibilității transportorului de dopamină striatală, în concordanță cu o scădere a integrității nigrostriatale (Figura 1). Un RMN de înaltă rezoluție a prezentat acum atrofia bilaterală a nucleului caudatus (Figura 3). Examenul neurologic a arătat un model discret de mers legat și hipomimie, dar nici o altă afecțiune a sistemului motor extrapiramidal și nici un simptom bulbar, cum ar fi distonia de hrănire. În afară de starea reflexă scăzută la nivelul membrelor inferioare, pentru care studiile de conducere nervoasă au confirmat o polineuropatie axonală senzorimotorie, examenul neurologic a fost normal. Simptomele neuropsihologice au inclus aspecte ale unei tulburări de anxietate, iar pacientul a raportat dificultăți cu pierderea memoriei pe termen scurt. Cu toate acestea, testarea neuropsihologică nu a confirmat defecțiunea mnestică vădită, ci mai degrabă o distragere nespecifică, care ar fi putut fi îmbunătățită suplimentar de suprimarea insuficientă a convulsiilor la momentul respectiv. Testele de laborator au fost negative pentru orice epilepsie simptomatică (i. e., anticorpi autoimune-encefalită, anticorpi antineuronali). Lichidul cefalorahidian nu a prezentat semne de inflamație, ci o creștere moderată a proteinelor (765 mg/l). Analiza aminelor biogene din lichidul cefalorahidian a relevat o creștere a glutaminei pe care am asociat-o cu crizele recente. În sângele periferic, s-au găsit 0,4% din acantocite. Testele serologice pentru boala Wilson au fost negative. Vizibil a fost o creștere persistentă a creatinkinazei (interval: 1.000–3.000 U/L) care a condus la diagnosticul suspectat de o tulburare mitocondrială. Pentru investigații ulterioare, a fost efectuat un test ischemic de lactat care a arătat rezultate normale. A fost efectuată o biopsie musculară a M. gastrocnemicus, dar analiza funcției mitocondriale și a lanțului respirator a fost normală.
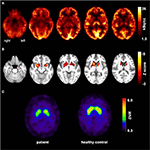
Figura 1. Rezultatele studiilor de imagistică moleculară. Imaginile transaxiale FDG-PET (A) au arătat nu numai un hipometabolism meziotemporal bilateral ușor (în conformitate cu originea convulsiilor meziotemporale), ci și un hipometabolism striatal marcat care s-a dovedit a fi foarte semnificativ în comparație cu controalele sănătoase . O examinare suplimentară FP-cit-SPECT (C) a evidențiat, de asemenea, o pierdere moderată a transportorilor de dopamină striatală, asistând la o scădere a integrității nigrostriatale (un control sănătos potrivit vârstei este prezentat pentru comparație; DVR, raportul volumului de distribuție).

Figura 2. Cursul clinic și diagnostic al pacientului index.
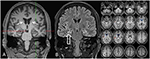
Figura 3. RMN prezintă atrofie discretă bilaterală a capului caudat și un hipocampus mai mic și hiperintens pe partea dreaptă, indicând scleroza hipocampală pe partea dreaptă . Hipocampul stâng este oarecum hiperintens, dar nu atrofic. Atrofia bilaterală a capului caudat este confirmată prin analiza CVR, în care culorile albastre arată scăderea culorilor gri și roșii creșterea volumului lichidului cefalorahidian, respectiv (C).
istoria familiei a dezvăluit o consangvinitate a părinților săi (veri primari), care ei înșiși nu aveau antecedente medicale semnificative. Dintre cei șase frați ai săi, doi (cu vârste cuprinse între 20 și 30 de ani) suferiseră până acum și de convulsii generale (vezi Figura 4). Dosarele medicale ale fraților săi nu au fost disponibile pentru noi.
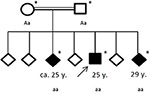
Figura 4. Pedigree al familiei afectate, cu vârsta primei crize epileptice în ani (y). Săgeată: index brevet. Asterisc: testarea genetică efectuată. Aa, heterozigot; aa, homozigot pentru c.4326 t>A (p.Tyr1442*).
sub ipoteza unei tulburări de mișcare neurodegenerativă ereditară, pacientul și familia sa au fost trimiși pentru teste genetice. Secvențierea exomilor a dezvăluit o mutație trunchiată homozigotă c. 4326 t>A (p.Tyr1442*) în gena ChAc VPS13A la cei trei frați afectați. Părinții consanguini au fost detectați heterozigoți. Nu au fost identificate alte variante sau mutații în secvențierea exomului.
de fapt, într-o urmărire la vârsta de 33 de ani, pacientul index a avut încă doar simptome extrapiramidale marginale și niciunul nu a fost prezent la niciuna dintre surorile sale. Acum a dat impresia unei ușoare rigidități doar sub amorsare pe membrul superior drept și o bradikinezie ușoară a membrelor superioare. În timpul tratamentului cu levetiracetam și lacosamid, pacientul nu a suferit crize convulsive.
discuție
în cazul de față, demonstrăm că ChAc se poate manifesta clinic cu epilepsie de lob temporal lipsită de simptome motorii semnificative datorită unei noi mutații corespunzătoare a genei VPS13A.
cursul bolii este caracterizat de obicei printr-o tulburare de mișcare progresivă (inclusiv Coreea, distonie, parkinsonism), modificări cognitive și psihiatrice și simptome miopatice cu markeri serologici de acantocitoză și hipercemie. Convulsiile au fost observate anterior ca un simptom al ChAc (6), dar tulburările de mișcare sunt încă considerate simptomul cheie. Există dovezi că epilepsia, și mai precis epilepsia originară din lobul temporal, ar putea fi un fenotip subestimat al ChAc. Peluso și colab. a raportat un pacient similar cu al nostru, care, chiar și la vârsta de 46 de ani, nu a prezentat tulburări de mișcare în timp ce prezenta constatări neuroimagistice indicative ale implicării ganglionilor bazali (7). În conformitate cu aceasta, Scheid și colab. au descris trei pacienți care au suferit de epilepsie și scleroză a lobului temporal mesial (pe baza studiilor RMN) ca simptom predominant și au fost diagnosticați cu ChAc (8). Mai mult, Mente și colab. a furnizat recent rezultatele autopsiei unui pacient ChAc care a prezentat nu numai atrofia ganglionilor bazali, ci și scleroza hipocampală (9). În conformitate cu cazul nostru, implicarea bilaterală a hipocampului pare a fi o constatare comună (10, 11). Relația exactă dintre convulsii și scleroză este, totuși, încă o chestiune de găină sau ou. Rolul corelant al corein în dezvoltarea structurală a hipocampului nu este încă pe deplin înțeles. Interesant, într-un model de șoarece de schimbare structurală ChAc a fost văzut, ca expresia proteinei hipocampale a receptorului GABA (a) gamma 2 și a proteinei sale de ancorare chorein au fost crescute (12).
în rutina clinică, acest fenotip poate fi încă o provocare în găsirea diagnosticului corect. Primul indiciu crucial spre o boală neurodegenerativă la pacientul nostru a fost o scădere proeminentă a metabolismului glucozei striatale și o scădere moderată a integrității nigrostriatale pe FDG-PET și, respectiv, FP-CIT-SPECT. Mica colecție de rezultate imagistice funcționale, bazate pe descrierea seriilor de cazuri, arată că alterările metabolismului și disfuncția dopaminergică apar mai ales în nucleul caudat și putamen (13). RMN-ul creierului a evidențiat atrofia nucleului caudatus în cursul bolii, care a fost descrisă ca o constatare tipică în ChAc (14). În consecință, caracteristicile neurologice distincte includ de obicei Coreea, distonia alimentară, diskineziile orofaciolinguale și ticurile, alături de alte simptome ale afecțiunii ganglionilor bazali, cum ar fi parkinsonismul și distonia (3). Este tentant să speculăm că modificările striatale observate la pacientul nostru erau încă sub pragul de a provoca simptome (adică descoperiri imagistice prodromale), care pot apărea în cele din urmă pe măsură ce boala progresează. Prin urmare, încurajăm imagistica structurală și moleculară ca instrument de diagnostic sensibil în această boală orfană.
în ChAc, există descrieri ale numărului de acantocite între 5 și 50% (3), dar există câteva rapoarte de caz care demonstrează că acantocitele pot apărea doar târziu în cursul bolii (15), pot fi foarte scăzute sau chiar absente complet (16). Prin urmare, acantocitele nu trebuie considerate obligatorii pentru efectuarea diagnosticului. În plus, epilepsia ar putea întârzia diagnosticul, deoarece deghizează alte simptome tipice ale ChAc. În special, simptomele psihiatrice, cum ar fi modificările de personalitate și deteriorarea cognitivă, care sunt foarte frecvente, pot fi interpretate greșit și atribuite efectelor secundare legate de medicamente (adică levetiracetam) sau legate de convulsii. Hipercemia secundară este observată după convulsii clonice tonice generale și o creștere persistentă ar putea fi trecută cu vederea.
cazul nostru evidențiază, de asemenea, beneficiul crucial al secvențierii exomilor pentru a stabili un diagnostic corect, în special în bolile rare sau atunci când simptomele sunt vagi. Familia genelor vps13 (A–D) de mamifere prezintă un interes în creștere, iar mutațiile au fost identificate în alte boli neurologice, cum ar fi boala Parkinson autosomală recesivă sau ataxia spinocerebelară autozomală recesivă (17). Mutațiile recesive în VPS13D provoacă tulburări de mișcare cu debut în copilărie (18). Fiziopatologia subiacentă a ChAc nu este încă descifrată pe deplin, dar s-a arătat că VPS13A codifică proteina coreină care este exprimată omniprezent în creier și într-un număr mare de alte țesuturi (19). Studiile arată că participă la căile de semnalizare care reglează arhitectura citoscheletală, exocitoza și supraviețuirea celulelor (20). O altă mutație care a fost descrisă pentru a prezenta un fenotip dominat de convulsii la 9 pacienți cu ChAc este mutația c.2343del în gena VPS13A (21). Este rezonabil să presupunem că mutațiile specifice din aceeași genă au ca rezultat o anumită aberație a coreinei care provoacă un fenotip unic, de exemplu prin afectarea diferitelor zone ale creierului. Cu toate acestea, fiziopatologia de bază este încă doar speculativă și documentația suplimentară a mutațiilor cauzale va fi de interes. Arătăm aici că mutația trunchiată c. 4326 t>A (p.Tyr1442*), care nu a fost descrisă anterior, are ca rezultat un fenotip similar cu epilepsie predominantă la toți membrii familiei afectate.
observații finale
epilepsia (în special epilepsia lobului temporal bilateral) este probabil să fie un fenotip predominant al ChAc. Prin urmare, trebuie efectuată cu atenție o căutare a steagurilor roșii suplimentare (cum ar fi hipercemia, istoricul familial, polineuropatia). În cazurile suspecte de căutare pentru acantocite în sânge și creier RMN ar trebui să fie adăugate ca aceste instrumente sunt disponibile pe scară largă. În caz de îndoială, diagnosticul trebuie completat cu FDG-PET și/sau FP-cit-SPECT, deoarece acestea sunt sensibile în detectarea implicării striatale. În epilepsiile autozomale recesive cu constatări indicative pentru o boală neurodegenerativă VPS13UN test unic de genă sau chorein Western blot ar trebui apoi utilizat pentru a stabili diagnosticul corect. Acestea din urmă ar trebui luate în considerare și în alte tulburări neurodegenerative fără etiologie definită genetic.
declarație de etică
consimțământul scris informat a fost obținut de la pacient pentru participarea la studiu. Consimțământul informat în scris a fost obținut de la pacient pentru publicarea acestui raport de caz.
contribuții ale autorului
JW, MR și SK au participat la managementul pacienților. JW a scris prima schiță a manuscrisului. LF, HU și PM au pregătit cifre. Toți autorii au contribuit la revizuirea manuscrisului, au citit și au aprobat versiunea trimisă.
Declarație privind conflictul de interese
HU este acționar al Veobrain Gmbh, un spin-off al Centrului Medical Universitar Freiburg.
autorii rămași declară că cercetarea a fost efectuată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
1. Walterfang M, Evans A, Leong Loi JC, Jung HH, Danek a, Walker RH și colab. Neuropsihiatria sindroamelor de neuroacantocitoză. Neurologi Biobehav Rev. (2011) 35:1275-83. doi: 10.1016/j.neubiorev.2011.01.001
Rezumat PubMed / CrossRef Text Complet / Google Scholar
2. Walker RH. Managementul sindroamelor de neuroacantocitoză. Tremor Alte Hiperkinet. Mov. (2015) 5:346. doi: 10.7916 / D8W66K48
PubMed rezumat / CrossRef Text Complet / Google Scholar
3. Velayos Baeza A, Dobson-Stone C, Rampoldi L, Bader B, Walker RH, Danek A și colab. Coreea-Acantocitoză. GeneReviews-ul. Seattle, WA: Universitatea din Washington (1993).
4. Critchley EM, Clark DB, Wikler A. Acantocitoză și tulburări neurologice fără Betalipoproteinemie. Arch Neurol. (1968) 18:134–40.
Rezumat PubMed
5. Levine sunt, Estes JW, Looney JM. Boala neurologică ereditară cu acantocitoză. Un nou sindrom. Arch Neurol. (1968) 19:403–9.
Rezumat PubMed / Google Scholar
6. Hardie RJ, Pullon HW, Harding AE, Owen JS, Pires M, Daniels GL și colab. Neuroacantocitoză. un studiu clinic, hematologic și patologic de 19 cazuri. Creier (1991) 114(Pt 1A):13-49.
Rezumat PubMed / Google Scholar
7. Peluso S, Bilo L, Esposito M, Antenora A, Rosa ADR, Pappata S, și colab. Coreea-acantocitoză fără coreeană: extinderea fenotipului clinic. Parkinson Legate De Disord. (2017) 41:124–6. doi: 10.2016/j.parkreldis.2017.05.013
PubMed Rezumat / CrossRef Text Integral / Google Scholar
8. Scheid R, Bader B, Ott DV, Merkenschlager a, Danek A. dezvoltarea epilepsiei lobului temporal mezial în Coreea-acantocitoză. Neurologie (2009) 73:1419-22. doi: 10.1212 / WNL.0b013e3181bd80d4
PubMed rezumat / CrossRef text integral / Google Scholar
9. Mente K, Kim SA, Grunseich C, Hefti MM, Crary JF, Danek A și colab. Scleroza hipocampală și epilepsia lobului temporal mezial în Coreea-acantocitoză: caz cu evaluare clinică, patologică și genetică. Neuropatol Appl Neurobiol. (2017) 43:542–6. doi: 10.1111 / nan.12403
Rezumat PubMed / CrossRef Text Complet / Google Scholar
10. Bader B, Vollmar C, Ackl N, Ebert A, la Foug Otrivre C, Noachtar S, și colab. Epilepsia bilaterală a lobului temporal confirmată cu EEG intracranian în Coreea-acantocitoză. Sechestru (2011) 20:340-2. doi: 10.1016 / j.sechestru.2010.12.007
PubMed Rezumat / CrossRef Text Integral / Google Scholar
11. Marson am, Bucciantini e, Gentile e, Geda C. Neuroacantocitoză: constatări clinice, radiologice și neurofiziologice într-o familie italiană. Neurol Sci. (2003) 24:188–9. doi: 10.1007 / s10072-003-0123-1
PubMed rezumat / CrossRef Text Complet / Google Scholar
12. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, și colab. Deficitul de coreină duce la reglarea în sus a gefirinei și a receptorului GABA(a). Biochem Biophys Res Comun. (2006) 351:438–42. doi: 10.1016 / j. bbrc.2006.10.070
PubMed Rezumat / CrossRef Text Integral / Google Scholar
13. Ehrlich DJ, Walker RH. Neuroimagistica funcțională și Coreea: o revizuire sistematică. J Clin Mov Disord. (2017) 4:8. doi: 10.1186 / s40734-017-0056-0
PubMed rezumat / CrossRef Text Complet / Google Scholar
14. Connolly BS, Hazrati L-N, Lang AE. Constatări neuropatologice în Coreea-acantocitoză: noi perspective asupra mecanismelor care stau la baza parkinsonismului și convulsiilor. Acta Neuropatol. (2014) 127:613–5. doi: 10.1007 / s00401-013-1241-3
PubMed rezumat / CrossRef Text Complet / Google Scholar
15. Sorrentino G, de Renzo a, Miniello s, Nori O, Bonavita V. apariția târzie a acantocitelor în cursul Coreei-acantocitoză. J Neurol Sci. (1999) 163:175–8.
Rezumat PubMed / Google Scholar
16. Bayreuther C, Borg M, Ferrero-Vacher C, Chaussenot A, Lebrun C. Coro-acantocitoză fără acantocite. Revue Neurol. (2010) 166:100–3. doi: 10.1016 / j. neurol.2009.03.005
CrossRef Text Integral / Google Scholar
17. Lesage S, Drouet V, Majounie e, Deramecourt V, Jacoupy M, Nicolas a, și colab. Pierderea funcției VPS13C în Parkinsonismul autosomal-recesiv determină disfuncție mitocondrială și crește mitofagia dependentă de PINK1/Parkin. Sunt Jum Genet. (2016) 98:500–13. doi: 10.1016 / j.ajhg.2016.01.014
PubMed Rezumat / CrossRef Text Integral / Google Scholar
18. Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M și colab. Mutațiile recesive în > VPS13D provoacă tulburări de mișcare cu debut în copilărie. Ann Neurol. (2018) 83:I6. doi: 10.1002 / ana.25204
Rezumat PubMed / CrossRef Text Complet / Google Scholar
19. Kurano Y, Nakamura M, Ichiba M, Matsuda M, Mizuno E, Kato M, și colab. Distribuția și localizarea in vivo a chorein. Biochem Biophys Res Comun. (2007) 353:431–5. doi: 10.1016 / j. bbrc.2006.12.059
PubMed Rezumat / CrossRef Text Integral / Google Scholar
20. Lang F, Pelzl L, Sch Oktifls l, Hermann A, F Oktifler M, Sch Oktiffer TE, și colab. Neuronii, eritrocitele și nu numai-diferitele funcții ale corein. Neurosignals (2017) 25:117-26. doi: 10.1159/000485457
PubMed rezumat / CrossRef Text Complet / Google Scholar
21. Benninger F, Afawi Z, Korczyn AD, Oliver KL, Pendziwiat M, Nakamura M și colab. Convulsiile ca simptom prezent și proeminent în Coreea-acantocitoză cu mutație genică c.2343del VPS13A. Epilepsia (2016) 57:549-56. doi: 10.1111 / epi.13318
PubMed Abstract | CrossRef Full Text | Google Scholar