frontiere în pediatrie
- Introducere
- sindromul QT lung
- Lqts1
- LQTS2
- LQTS3
- LQTS4
- LQTS5
- LQTS6
- LQTS7
- LQTS8
- Lqts9 și LQTS10
- LQTS11
- Lqts12
- LQTS13
- LQTS dobândite
- caracteristicile electrocardiografice în cele trei forme comune ale sindromului QT lung
- genotip-fenotip
- managementul clinic al LQTS
- : Perspectiva Saudită
- Declarație privind conflictul de interese
- mulțumiri
Introducere
aritmiile cardiace familiale sau ereditare cuprind procente semnificative de aritmii și, de asemenea, cauzale morții subite cardiace (SCD) (1, 2). În ultimele două decenii, oamenii de știință și clinicienii au depus eforturi enorme pentru a descoperi mecanismele complicate și complexe ale aritmiilor familiale congenitale (1-8). Pentru a înțelege mecanismul aritmogenezei, trebuie să cunoaștem elementele de bază ale structurii celulare cardiace și proprietățile lor electrofiziologice. Miocitele cardiace sunt principalele celule funcționale din inimă și sunt cuplate extensiv, astfel încât impulsurile se propagă rapid și uniform. Cardiomiocitele sunt separate una de cealaltă printr-o limită specializată numită disc intercalat; proteinele de joncțiune gap, desmosomii cardiaci și canalele ionice sunt localizate în discul intercalat (9). Joncțiunile Gap constau din conexiuni bine ambalate care permit schimbul intercelular de molecule mici și, de asemenea, permit curgerea curenților excitatori de la o celulă la celula vecină. Desmosomii împreună cu joncțiunile adherens sunt responsabili pentru atașamentele mecanice ale cardiomiocitelor individuale. Toate aceste componente din discurile intercalate sunt segregate secvențial și fiecare componentă își exercită funcția unică; întreruperea componentei unice afectează funcția altor componente, ceea ce predispune inima să dezvolte aritmii (9-11). Canalele ionice cardiace sunt complexe proteice care formează pori care asigură o mișcare de tensiune închisă și coordonată în mod complicat spre interior și spre exterior a curenților ionici de-a lungul membranelor celulare, esențiale pentru generarea și propagarea ritmului cardiac. Sindromul QT lung( LQTS), sindromul QT scurt (SQTS), sindromul sinusului bolnav (SSS), defectul de conducere cardiacă (CCD), BrS, tahicardia ventriculară polimorfă catecolaminergică (CPVT), sindromul de repolarizare precoce (ERS) și fibrilația atrială familială (AF) sunt canalele cardiace cunoscute în prezent, care ar putea apărea din cauza unui defect unic sau multiplu al genelor legate de generarea și propagarea ritmului cardiac.
în această revizuire, vom descrie exclusiv aritmiile legate de LQTS, fiziopatologia și managementul clinic Disponibil în prezent. Am fost pionieri în elucidarea patologiei genetice în aritmiile cardiace familiale în Arabia Saudită (1, 12-14), încă efectuăm investigații cardiogenetice în Arabia Saudită, la sfârșitul acestei revizuiri, vom discuta despre cunoștințele disponibile în prezent despre constatările genetice și clinice obținute de la mai multe familii din Arabia Saudită cu antecedente de sincopă și SCD. Vom adăuga, de asemenea, datele publicate recent despre LQTS de la o echipă de la Spitalul de specialitate King Faisal și Centrul de cercetare, Riyadh (15).
sindromul QT lung
LQTS Congenital este o afecțiune moștenită definită prin prelungirea intervalului QT pe electrocardiogramă (ECG). Pacienții cu toate formele de LQTS sunt predispuși la tahiaritmia ventriculară, torsada vârfurilor (TdP) care duce la sincopă recurentă sau SCD. În multe cazuri, sincopa sau moartea subită ar putea fi prima și singura manifestare. LQTS afectează aproximativ 1 din 2.000 de persoane din întreaga lume (16). Semnul distinctiv al LQTS este prelungirea intervalului QT pe ECG (corectat pentru ritmul cardiac, adică QTc). Valorile normale ale QTc sunt de 440 ms la bărbați și 450 ms la femei. La copii, valorile dependente de vârstă și sex sunt relevante. Recomandarea recentă de consens pentru diagnosticul LQTS este următoarea (17, 18):
1. LQTS este diagnosticat:
a. în prezența unui scor de risc LQTS 3.5, în absența unei cauze secundare de prelungire a intervalului QT și/sau
b. în prezența unei mutații patogene neechivoc în una dintre genele LQTS sau
c. În prezența unui interval QT corectat pentru frecvența cardiacă utilizând formula Bazett (QTc), 500 ms, în electrocardiogramă repetată cu 12 plumb și în absența unei cauze secundare de prelungire a intervalului QT.
2. LQTS poate fi diagnosticat în prezența unui QTc între 480 și 499 ms în ECG repetate cu 12 plumb la un pacient cu sincopă inexplicabilă în absența unei cauze secundare de prelungire a QT și în absența unei mutații patogene.
pacienții cu durată QTc 500 ms au avut o probabilitate cumulativă semnificativ mai mare de a prezenta prima lor sincopă comparativ cu pacienții cu durată QTc <500 ms de la naștere până la vârsta de 20 de ani (19). Dar, la pacienții care au prezentat 2, 3 și 4 episoade de sincopă, riscul unui episod ulterior de sincopă a fost practic identic între pacienții care au avut o durată QTc îngustă sau prelungită (19). Baza moleculară a LQTS este eterogenă și până în prezent, mutațiile din 13 gene diferite au fost descrise cauzal la LQTS (1-8, 19, 20). Un defect genetic se găsește de obicei la 70% dintre pacienții cu LQTS într-una din aceste 13 gene (1-8, 19, 20). Dintre cele 13 tipuri diferite de LQTS cunoscute în prezent, cele mai frecvente sunt LQTS1, LQTS2 și lqts3, datorită defectelor genelor canalului ionic cardiac, KCNQ1, KCNH2 și, respectiv, SCN5A. Nouăzeci la sută din toate mutațiile cauzale LQTS se găsesc în aceste trei gene (1-8, 19, 20). Mutațiile din restul de 10 gene sunt rare și cuprind doar 10% din toate mutațiile LQTS cunoscute în prezent.
sindromul QT lung este de obicei o boală autozomală dominantă, dar, ocazional, mutații multiple într-o singură genă sau în gene diferite ar putea fi găsite la 5-10% dintre pacienții cu LQTS (21, 22). Pacienții cu mutații multiple ar putea prezenta un QTc mai lung în comparație cu cei cu o singură mutație și acești pacienți prezintă, de asemenea, un risc crescut de 3,5 ori mai mare pentru evenimente cardiace care pun viața în pericol(21, 22)
Lqts1
mutațiile genei KCNQ1 sunt cea mai comună formă a tuturor LQTS și se referă la LQTS de tip 1 (LQTS1). Cincizeci la sută din toate mutațiile cauzale LQTS se găsesc în gena KCNQ1 (20). KvLQT1 (numit și Kv7.1) este o proteină produsă de gena KCNQ1 și proteina formează tetrameri în reticulul endoplasmatic din interiorul celulelor, care se co-asamblează putativ cu nurca proteică (codificată de KCNE1) și sunt apoi transportați în membrana plasmatică a miocitelor cardiace, unde mediază un curent activator lent care accelerează repolarizarea potențialului de acțiune în țesuturile cardiace, acest curent este cunoscut sub numele de IKs (Figura 1). Mutațiile cauzale lqts1 kcnq1 sunt în mare parte mutații missense și, în cazuri rare, ar putea fi mutații frameshift în regiunea C-terminală (23). Aritmia cardiacă la purtătorii de mutații KCNQ1 este declanșată de unități adrenergice, de exemplu, stres emoțional, efort fizic, scufundări, înot (24-26). Pacienții cu mutații la domeniile transmembranare ale KvLQT1 prezintă un risc mai mare de evenimente cardiace legate de LQTS și au o sensibilitate mai mare la stimularea simpatică (26, 27). Pacienții cu mutații missense prezintă un risc crescut comparativ cu pacienții cu mutații fără sens sau trunchiere (27, 28). Variația 3 ‘ – UTR în gena KCNQ1 afectează, de asemenea, susceptibilitatea aritmiei în mod semnificativ, probabil prin impactul asupra expresiei genei (29). Pacienții cu aritmii cauzate de mutațiile KCNQ1 răspund destul de bine la blocanții de la blocante, dar unii pacienți ar putea fi în continuare mai puțin receptivi sau chiar rezistenți la acest medicament. Într-un articol recent, Barsheshet și colab. (30) a susținut că pacienții cu mutații în afara regiunii buclei citoplasmatice (bucla c) din KvLQT1 sunt mai puțin receptivi la blocanții de la XV.
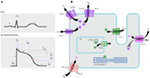
Figura 1. Curenții ionici care contribuie la potențialul de acțiune ventriculară (a) și Reprezentarea schematică a unui cardiomioc care prezintă (numai) acele proteine implicate în patogeneza sindroamelor de aritmie moștenită (b). În (A), potențialul de acțiune este aliniat cu timpul său aproximativ de acțiune în timpul ECG. În (B), ankyrin-B, o proteină adaptor implicată în sindromul QT lung de tip 4, nu este descrisă.
mutațiile heterozigote homozigote sau compuse în gena KCNQ1, care ar putea provoca forma recesivă a bolii, sindromul Jervell și Lange-Nielsen (JLNS), de tip 1 (31), sunt destul de rare. Pacienții cu JLNS suferă de aritmii cardiace severe și surditate (31, 32). Pacienții cu mutații kcnq1 cauzale la JLNS nu au de obicei IKs funcționale (28, 31-33). Este posibil ca unii pacienți să nu aibă surditate în ciuda faptului că au mutații heterozigote homozigote sau compuse în KCNQ1, astfel de cazuri sunt denumite lqts1 autosomal recesiv (13, 32). La acești pacienți, o cantitate mică de curent funcțional IKS (< 10% din totalul IKs) ar putea fi încă prezentă, ceea ce menține funcția auditivă, dar defectele lor de ritm cardiac sunt la fel de severe ca la pacienții cu JLNS (13, 32).
LQTS2
acest tip de LQTS este la fel de răspândit ca LQTS1, reprezentând 35-40% dintre pacienții cu LQTS cu o mutație detectabilă(1, 20, 24, 25). Kcnh2 codifică pentru proteina HERG (Kv11.1), Care este subunitatea X-X a curentului k+ (IKr) al redresorului întârziat cu activare rapidă. Mutațiile patogene din această genă care reduce funcția canalului Kv11.1 prelungește durata intervalului QT (Figura 1) și sunt cauzale la LQTS2. Douăzeci și nouă la sută din atacurile sincopale din LQTS2 apar în timpul odihnei/somnului și doar 13% din atacurile sincopale au fost raportate să apară în timpul exercițiilor fizice (25, 34). Zgomotele bruște uimitoare, de exemplu, inele de ceas deșteptător, sonerii, inele telefonice, declanșează de obicei atacuri sincopale la acești pacienți (24, 34). Pacienții cu mutații în regiunea de formare a porilor Kv11.1 (codificată de gena KCNH2) sunt susceptibili la risc crescut de evenimente cardiace legate de aritmie, comparativ cu pacienții cu mutații din regiunea non-porilor (35). La nou-născuți, blocul Atrioventricular 2:1 (AV) este asociat preferențial cu mutații KCNH2 (36). Blocul AV complet complicat de LQTS a fost, de asemenea, găsit la 17% dintre pacienții adulți cu o mutație a genei KCNH2 (37). Mutațiile homozigote în KCNH2 sunt rare și atunci când sunt prezente, pacienții suferă de o formă severă de LQTS, cu 2:1 bloc AV și aritmii ventriculare severe, atât în stadiile intrauterine, cât și după naștere (11, 38-40). În plus față de numeroasele mutații descrise în patologia LQTS2, variantele polimorfe comune din gena KCNH2 ar putea modula, de asemenea, severitatea bolii. Un exemplu interesant este polimorfismul K897T (SNP) în KCNH2, care este prezent în 33% din populația generală (41). S-a raportat că K897T exacerbează patogenitatea mutațiilor kcnh2 (42, 43).
LQTS3
Nav1.5 este subunitatea de formare a porilor din canalul cardiac na+ dependent de tensiune, este o proteină membranară integrală codificată de gena SCN5A și este implicată în inițierea și conducerea potențialelor de acțiune cardiacă. Canalele cardiace na + sunt compuse dintr-o subunitate de formare a porilor (subunitate codificată de SCN5A) și una sau mai multe subunități auxiliare de formare a porilor.
LQTS3 este cauzat de amplificarea mutațiilor funcționale care perturbă inactivarea rapidă a subunității de tip VIII (Figura 1) și o mutație a genei SCN5A a fost descrisă la < 10% din toți pacienții cu LQTS cu o mutație (1, 20, 24). Pacienții cu LQTS3 prezintă majoritatea (39%) evenimentelor cardiace în timpul somnului/odihnei (25) și aproximativ 13% dintre evenimente au fost raportate în timpul exercițiului fizic (25). În mai multe cazuri, s-a demonstrat că o singură mutație SCN5A exercită două sau chiar trei fenotipuri distincte de aritmii în aceeași familie, de exemplu, LQTS, BrS sau CCD (44-47). Pacienții de sex masculin cu o mutație LQTS3 ar putea dezvolta simptome mult mai devreme decât pacienții de sex feminin (48). Mutațiile scn5a heterozigote și homozigote au fost descrise în LQTS3 cu bloc AV funcțional 2:1 (49). Am găsit recent o familie iraniană cu mutația 1507_1509delqkp la mai mulți membri ai familiei, unde pacienții au combinat LQTS și CCD (datele nu sunt prezentate). Această mutație a fost raportată și la pacienți din alte țări, ceea ce sugerează că aceasta este o mutație recurentă și punct fierbinte . Mutațiile cu astfel de caracteristici de pierdere și câștig de funcție în timpul diferitelor faze ale potențialului de acțiune au fost, de asemenea, raportate în mutațiile 1493delk și 1795insd (44, 51).
ocazional, un SNP ar putea exercita, de asemenea, un efect patogen asupra purtătorilor săi. S1103Y este o variantă comună în gena SCN5A, prezentă la 13% dintre afro-americani (52). Purtătorii cu această variantă prezintă un risc crescut de aritmii și sindrom de moarte subită a sugarului (SIDS) (52).
LQTS4
LQTS4 reprezintă prima formă non-canal de LQTS. O mutație în ANK2, o proteină adaptoare, duce la supraîncărcarea intracelulară a calciului care contribuie la LQTS4 (53, 54). În plus față de prelungirea QT, pacienții cu acest sindrom ar putea avea bradicardie sinusală, Af paroxistică și CPVT (54). Efectul patogen al mutațiilor ANK2 poate fi moderat până la sever, iar expresiile clinice depind de severitatea mutației.
LQTS5
mutațiile KCNE1 sunt asociate cu Lqts5 (Figura 1) (55, 56). Mutațiile heterozigote kcne1 reduc IKs prin exercitarea unui efect negativ dominant asupra alelei normale însoțitoare și duc la repolarizarea cardiacă întârziată (Figura 1), responsabilă de riscul crescut de aritmii (6). Pacienții cu mutații homozigote kcne1 suferă de JLNS (tip 2) (57, 58).
D85N este un polimorfism în gena KCNE1, prezent în 0.7-1% din populația generală (41). Într-un studiu realizat de Nishio și colab. (59), polimorfismul D85N a fost mai frecvent întâlnit la pacienții cu LQTS, făcându-l un genotip de risc în patologia LQTS (potențial doar la populația asiatică). În Europa, D85N a fost raportat la 5% dintre pacienții cu LQTS dobândite (aLQTS) (într-o cohortă de 32 de pacienți), care au prezentat TdP (60).
LQTS6
gena KCNE2 codifică pentru peptida 1 legată de nurcă (MiRP1), o subunitate presupusă a canalului cardiac de potasiu IKr (Figura 1). Mutațiile genei KCNE2 ar putea duce, de asemenea, la defecte ale componentei cu activare rapidă a curentului de potasiu redresor întârziat (IKr), baza patologică a LQTS6 (61). Stimul auditiv / acustic, cum ar fi zgomotul ceasului deșteptător, soneria etc. ar putea provoca atacuri sincopale la purtătorii de mutații KCNE2, similar cu mutația KCNH2 (62).
LQTS7
acest sindrom este, de asemenea, cunoscut sub numele de sindromul Andersen–Tawil (ATS). ATS este o tulburare rară, manifestată prin sincopă ocazională și stop cardiac. Caracteristicile ECG includ prelungirea ușoară a intervalului QT, undele u anormale, ectopia ventriculară frecventă, tahicardia ventriculară bidirecțională (VT) și Vt polimorfă. Acest sindrom prezintă, de asemenea, caracteristici extracardiace, de exemplu, paralizia periodică a mușchilor scheletici și probleme de dezvoltare, cum ar fi Palatul despicat, urechile joase, statura scurtă și defectele de dezvoltare ale membrelor (63). Majoritatea pacienților diagnosticați clinic cu ATS au raportat o mutație a KCNJ2 (63). KCNJ2 codifică o subunitate de formare a porilor de rectificare interioară a canalelor de potasiu (IK1) (Figura 1) (64, 65).
LQTS8
de asemenea cunoscut sub numele de sindromul Timothy (TS), pacienții prezintă o prelungire severă a QT pe ECG-urile lor, care este combinată cu sindactilie, chelie la naștere și dinți mici în 100% din cazuri și malformații structurale cardiace mai puțin penetrante, retard mental, autism și caracteristici dismorfice faciale (66). Există două subtipuri: TS1 (clasic) și TS2 (formă rară).
TS2 este mai sever din punct de vedere cardiologic decât TS1 (66, 67). De asemenea, pacienților cu TS2 le lipsește sindactilia (67). Mutațiile din subunitatea XV-1 a curentului de calciu de tip L (ICa-L) care codifică gena CACNA1C conduc la ambele forme de TS (LQTS8). Există Exon-8 alternativ îmbinate, care se exclud reciproc în gena CACNA1C, pentru claritate, ele sunt denumite exon-8 și exon-8A. în inimă și creier, unde CACNA1C este exprimat predominant, exon-8 se găsește în 80% din transcrierile ARNm din hectolitru, iar exon-8A este prezent în transcrierile 20% din ARNm (66). Mutația G406R în exon-8A este cauzală formei clasice de TS (TS1) și G402S în exon-8 a fost raportată în severer în TS2. O nouă adăugare la această listă este mutația Ala1473Gly, care a fost descrisă la un sugar TS cu fenotip extins suplimentar (68). Toate mutațiile au fost fie de novo, fie mozaic la părinte și toate rezultatele câștigului funcției canalului ICa-L (66-69).
Lqts9 și LQTS10
mutațiile CAV3 sau SCN4B produc câștig de funcție la INa târzie, provocând un fenotip asemănător LQTS3 (70-74). Acestea sunt cunoscute sub numele de lqts9 (asociat cu mutația CAV3) și LQTS10 (asociat cu mutația SCN4B).
Caveolae sunt frumos descrise de Engelman și colab. (72) ca “peșteri mici” în membrana plasmatică. Acestea sunt mici gropi neacoperite și este considerat ca site – ul de importante evenimente dinamice și de reglementare la membrana plasmatică (72, 73). Caveolinele sunt principalele proteine din caveolae, iar caveolina – 3 (codificată de gena CAV3) se găsește în mod specific în cardiomiocite și celulele musculare scheletice. Mai multe canale ionice cardiace au fost raportate în mod specific ca fiind localizate în caveolele extrase din miocitele cardiace care sunt îmbogățite În caveolin-3 (72, 73). În plus, componentele cascadei de semnalizare a receptorului-adrenergic al receptorului-adrenergic sunt prezente și în membranele îmbogățite cu caveolae (72, 73).
SCN4B codifică pentru Nav XV4, care este o subunitate auxiliară XV a canalului cardiac de sodiu. Până în prezent, o singură mutație (L179F) a fost raportată în această genă într-o familie mexicană cu mai mulți membri ai familiei afectate (74). S-a constatat că mutația acestei gene are ca rezultat câștigarea funcției curentului Nav1.5 (74).
LQTS11
în inimă, reglarea simpatică a duratei potențialului de acțiune cardiacă (APD) este mediată de activarea receptorului-adrenergic (XV-AR), care necesită asamblarea AKAP9 (Yotiao) cu subunitatea-XV (KvLQT1) a canalului IKs. Mutația în AKAP9 provoacă LQTS11 (75). Până în prezent, a fost raportată o singură mutație, S1570L, în AKAP9 (75).
Lqts12
mutația genei-1-Sintrofinei (SNTN1) este cauzală pentru lqts12. Mutația în această genă duce la câștigarea funcției canalului cardiac de sodiu (Nav1.5), care este baza patologică a LQTS12 (76).
LQTS13
subunitatea canalului de potasiu cuplată cu proteine G, rectificatoare în interior (Kir3.4) este codificată de gena KCNJ5. O mutație a pierderii funcției în această genă ar putea provoca LQTS13 (77). Până în prezent, o singură mutație, G387R, a fost descrisă într-o familie chineză cu nouă pacienți care au această mutație. Expresia redusă a membranei plasmatice a Kir3.4 a fost sugerată ca patologie a LQTS la pacienți.
LQTS dobândite
în plus față de LQTS congenitale, există și o altă variantă a LQTS cunoscută sub numele de aLQTS, care este cauzată de factori și substanțe care scad fluxul de potasiu și afectează capacitatea miocardului de a repolariza. Condițiile bine recunoscute sunt sexul feminin, hipokaliemia și medicamentele care inhibă canalele cardiace de potasiu (78-80). Un număr de medicamente prescrise în mod obișnuit ar putea, de asemenea, să lege și să blocheze canalul HERG (Kv11.1, o proteină codificată de gena KCNH2) și predispune la aLQTS (79, 80). Recent, sa demonstrat că blocarea IKs ar putea contribui, de asemenea, la aLQTS induse de medicamente, în special atunci când Rezerva de repolarizare este compromisă (81). S-a constatat că fluoxetina și norfluoxetina suprimă proprietățile IKs, atât in vivo, cât și in vitro și au condus la LQTS marcate (81). Polimorfismele, D85N în nurcă (gena KCNE1), T8A, Q9E în MiRP1 (gena KCNE2), care sunt presupuse subunități-subunități ale canalelor IKS și IKr, au fost raportate pentru a provoca aLQTS (82, 83). De asemenea, s-a raportat LQT autoimune la un pacient cu IgG care conține anticorpi anti-HERG (84).
caracteristicile electrocardiografice în cele trei forme comune ale sindromului QT lung
tiparele tipice de undă ST-T sunt prezente la majoritatea pacienților cu LQTS genotipate și pot fi utilizate pentru identificarea genotipurilor LQTS1, LQTS2 și, eventual, lqts3 (85, 86).
forma LQTS1 a LQTS este asociată cu o undă t largă fără scurtarea intervalului QT la exercițiu (figura 2a). LQTS2 este asociat cu unde T de amplitudine mică, adesea bifide (figura 2b). LQTS3 este asociat cu un segment iso-electric lung și o undă t înaltă, îngustă (figura 2c). Dependența de pauză a debutului TdP în LQTS congenitale este specifică genotipului, fiind predominantă în LQTS2, dar aproape absentă în LQTS1 (87).

Figura 2. Înregistrări electrocardiografice de la pacienți cu LQTS1, LQTS2 și lqts3. (A) ECG cu 12 plumb al unui bărbat de 18 ani cu o mutație KCNQ1. Intervalul QT este prelungit(QTc = 500 ms). Segmentul ST are o bază largă și o amplitudine relativ mare. Intervalul de conducere este normal (calibrare standard). (B) ECG cu 12 plumb al unei fete de 14 ani cu mutație KCNH2. Intervalul QT este prelungit (QTc-520 ms). Segmentul ST este crestat în plumb V3 și are o amplitudine relativ scăzută în conductele de extremitate. Intervalul de conducere este normal (calibrare standard). (C) ECG cu 12 plumb al unui băiat de 12 ani cu mutație SCN5A. Intervalul QT este prelungit (QTc-600 ms). Segmentul ST are un segment izoelectric lung (aproape) cu o undă T mare, ascuțită și îngustă. Intervalul de conducere este normal (calibrare standard).
deși tiparele pot sugera un genotip specific al LQTS, s-au găsit frecvent excepții.
genotip-fenotip
sindromul QT lung este o boală autozomală dominantă. Analiza genotipului și fenotipului în rândul purtătorilor de mutații heterozigote a fost efectuată destul de extensiv la pacienții cu lqts1, LQTS2 și lqts3. În timpul copilăriei, riscul de evenimente cardiace este semnificativ mai mare la bărbații cu LQTS1 decât la femeile cu lqts1, în timp ce nu s-au observat diferențe semnificative legate de sex în ceea ce privește riscul de evenimente cardiace la pacienții cu LQTS2 și LQTS3 (88-91). În timpul maturității (și după vârsta de 40 de ani), femeile LQTS1 și lqts2 ar putea avea un risc semnificativ mai mare de evenimente cardiace decât bărbații respectivi (88-91). În general, letalitatea evenimentelor cardiace pare să fie predominantă la pacienții cu LQTS3 decât la pacienții cu LQTS1 și lqts2 (91). Femeile cu LQTS au un risc redus de evenimente cardiace în timpul sarcinii, dar riscul crește destul de mult în perioada postpartum de 9 luni, în special la femeile cu mutație în gena KCNH2 (92).
moartea subită cardiacă la copii ar putea fi cauzată și de mutații ale genelor canalului ionic cardiac (93-96). Aproximativ 28% dintre copiii cu un SCD inexplicabil (între 1 și 18 ani, vârsta medie: 12,3 3,8 ani) au fost purtători de mutații în genele cauzale LQTS (97). În SIDS, mutațiile în SCN5A păreau predominante (98, 99), dar au fost găsite și mutații în KCNQ1, KCNH2, KCNE2 și CAV3, SCN4B și SCN3B (100, 101). De asemenea, au fost raportate decese fetale Intrauterine din cauza defectelor genelor canalului ionic cardiac (12, 102).
managementul clinic al LQTS
încetarea tuturor medicamentelor despre care se știe că prelungesc intervalul QT și, de asemenea, corectarea dezechilibrelor electrolitice și/sau precipitarea condițiilor metabolice ar trebui să fie accentul principal în timpul tratamentului pacienților cu LQTS (dobândite). Simptomele la LQTS sunt adesea mediate adrenergic, de aceea se recomandă în general restricționarea participării pacienților la activități sportive (24, 25). Pilonul principal al terapiei clinice pentru LQTS este blocada de la XV. Preparatele cu acțiune lungă de propranolol, nadolol și metoprolol sunt de obicei utilizate, iar eficacitatea lor în blocada de la XV este evaluată prin tocirea ritmului cardiac al exercițiului (de exemplu, prin >20%) (24, 25). Dintre toate blocantele de la Centauri, propranololul și nadololul sunt considerate superioare metoprololului la pacienții simptomatici (103). În plus, blocada de la XV poate fi utilizată și ca tratament profilactic în purtătorii de mutații silențioase pentru a reduce SCD (24). Într-un studiu realizat de Barsheshet și colab. (30), pacienții cu mutații missense c-loop în gena KCNQ1 au prezentat un risc ridicat de evenimente cardiace care pun viața în pericol și au avut beneficii semnificative din tratamentul cu blocante de blocante ale centaurilor. Deoarece femeile cu LQTS2 prezintă un risc crescut în timpul perioadei postpartum de 9 luni, blocantele de circulație a sângelui trebuie prescrise pentru a reduce orice evenimente cardiace în această perioadă cu risc crescut (92). Un cardioverter–defibrilatoare implantabile (ICD) poate fi luat în considerare pentru pacienții cu sincopă recurentă în ciuda terapiei cu blocante de la XV sau la pacienții cu risc crescut de stop cardiac (de ex., lqts2 simptomatic și LQTS3 cu o prelungire documentată a QTc). Denervarea simpatică cardiacă stângă (LCSD) este recomandată pacienților cu LQTS cu risc ridicat la care un ICD este contraindicat sau refuzat, iar blocantele cu Centauri nu sunt eficiente, nu sunt tolerate, nu sunt acceptate sau contraindicate (18, 104). Pacienții cu JLNS au, de obicei, un QTc >500 ms și sunt, de asemenea, cu risc crescut, blocanții cu Centauri au eficacitate insuficientă la acești pacienți la care a fost recomandată o terapie precoce cu ICD (18, 31).
: Perspectiva Saudită
primul raport privind LQTS din Arabia Saudită a fost publicat în 1993 de la Spitalul Forțelor Armate din Riyadh (105). Patru sugari și copii mici între 6 și 48 de luni cu antecedente de convulsii recurente, dintr-o singură familie, au fost diagnosticați ca LQTS (105). Istoria familiei a arătat că alți doi membri ai familiei extinse au avut episoade similare de pierdere bruscă a conștiinței și trei membri ai familiei au murit brusc (105). În toate cazurile, diagnosticul inițial a fost epilepsia (105). Câțiva ani mai târziu, au fost raportate două rapoarte de caz sporadice cu varianta relativ mai severă a LQT-urilor neonatale combinate cu blocul AV 2:1 (106, 107). Toate aceste rapoarte clinice publicate au fost fără descoperiri genetice care ar putea explica fiziopatologia LQTS în ele (105-107).
noi, pentru prima dată, am raportat defecte genetice ca bază patologică a LQTS la pacienți similari din Arabia Saudită. Am investigat șase familii Saudite cu antecedente de sincopă și decese subite inexplicabile ale fătului, nou-născuților și copiilor (12-14). Autozomal recesiv LQTS1 a fost diagnosticat la copii din două familii (Figura 3). Autozomal recesiv LQTS2 a fost diagnosticat în două familii (Figura 4). Într-o familie, o pacientă de sex feminin a fost diagnosticată cu LQTS autosomal dominant 2 (Figura 5), pacientul a avut atacuri sincopale în timpul perioadei de recuperare postpartum la spital, ceea ce este foarte frecvent la femeile purtătoare de mutații KCNH2. La toți pacienții noștri, identificarea mutațiilor patogene în genele cauzale ale canalului ionic cardiac LQTS a condus la un diagnostic clinic confirmat, care au fost diagnosticate greșit ca convulsii epileptice înainte de a fi trimise la noi (13, 14) recent, Shinwari și colab. (15) de la Spitalul de specialitate King Faisal și Centrul de cercetare au raportat o mutație cauzală lqts1 kcnq1, H258P, într-o familie numeroasă cu 12 persoane afectate. Doar doi purtători au fost simptomatici, QTc-ul lor a fost >500 ms, iar blocantele de valori ale centicentrului au suprimat simptomele clinice la un pacient, iar la cel de-al doilea pacient simptomatic a fost necesară o ICD.
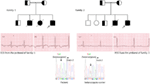
Figura 3. Desenul Pedigree al familiei-1 și 2 cu LQTS autosomal recesiv1, probandii sunt arătați cu o săgeată. ECG-urile din probands în două familii sunt prezentate în mijloc, cu un QTc de 557 și, respectiv, 529 ms. Mutația intronică, c. 387 -5 t > A în gena KCNQ1 a fost găsită la pacienți (prezentată în partea de jos). Mutația a fost arătată cu o săgeată. Cercurile și pătratele neumplute nu sunt purtătoare pentru mutație. Persoanele afectate sunt prezentate ca cercuri umplute (feminin) și pătrate (masculin). Pătratele și cercurile pe jumătate umplute sunt indivizi cu mutație heterozigotă. Persoanele decedate sunt indicate prin tăieturi, probandii sunt indicați printr − o săgeată, iar căsătoria consanguină este indicată prin = limita Exon-intron este indicată printr-o linie punctată cu săgeata îndreptată spre exon.
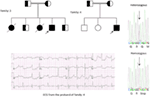
Figura 4. Desenul Pedigree al familiei-3 și 4 cu LQTS autosomal recesiv2. ECG al probandului din family-4 este prezentat sub desenul pedigree, care prezintă tahicardie sinusală, bloc AV aproape complet, ritm de evacuare complex, cu intervale QTc foarte lungi (QT >600 ms). Mutația, c.3208 c > T (P.Q1070X) în gena KCNH2 a fost găsită la pacienți (prezentată în dreapta), marcată cu o săgeată.
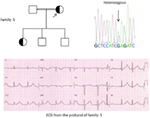
Figura 5. Stânga sus: pedigree al familiei-5. Individul afectat este prezentat de Cercul umplut (feminin) Proband este indicat de o săgeată. 12-ECG de plumb al probandului (I:2). ECG prezintă un val T de amplitudine mare, larg, cu amplitudine mare, intervalul QTc este de 580 ms. screeningul genei KCNH2 arată substituția nucleotidei “G” pentru un “A “(C. 2362g > a, săgeată marcată), ceea ce duce la substituția aminoacizilor, p.e788k.
constatările genetice și clinice din studiul nostru (12-14) din Arabia Saudită sunt destul de interesante din mai multe motive: (1) în total, am investigat șase familii, dintre care patru au fost homozigote/heterozigote compuse pentru mutații, iar mutațiile au provenit dintr-o sursă ancestrală; (2) toate mutațiile în LQTS au fost noi, raportate doar în aceste familii arabe; (3) datorită homozigozității sau heterozigozității compuse pentru mutații, fenotipurile clinice au fost severe și în familiile studiate (12-14). Am sugerat că observațiile genetice și fenotipice au rezultat din rata extrem de ridicată a căsătoriilor consanguine din Arabia Saudită (108, 109). Studiul nostru a furnizat primele dovezi științifice despre rolul consangvinității în exercitarea unui rol esențial în aritmii cardiace inexplicabile și SCDs la copii și adolescenți din Arabia Saudită (12-14). De asemenea, am arătat că mutația KCNQ1 c.387 -5 T > A (Nm_000218) (Figura 3) s-a răspândit în provincia Assir din Arabia Saudită de la un strămoș comun pe parcursul mai multor generații datorită incidenței ridicate a căsătoriilor consanguine . Până în prezent, aceasta este cea mai frecventă mutație cauzală LQTS1 în populația Arabiei Saudite, care a fost observată și la Spitalul specializat King Faisal și la Spitalul Militar Khamis Mashayt (Nepublicat). Un rezultat similar a fost obținut și pentru mutația cauzală lqts2, c. 3208 c > T (p.Q1070X) (Figura 4) în gena KCNH2 (12, 14), care este, de asemenea, o mutație fondatoare în Arabia Saudită și segregată de-a lungul multor generații în regiunea Assir (Vezi harta, Figura 6). Am prezice că există un număr considerabil de indivizi cu mutațiile ancestrale/fondatoare menționate (atât în KCNQ1, cât și în kcnh2 și alte gene) în această regiune și, de asemenea, în marile orașe precum Riyadh, Jeddah și Dammam din cauza migrației urbane. Mai multe mutații fondatoare sau ancestrale patogene pentru LQTS sunt foarte mult avute în vedere în alte provincii din Arabia Saudită datorită ratei ridicate a căsătoriilor consanguine.
Figura 6. Harta Arabiei Saudite (curtoazie: Wikipedia). Regiunea Assir este marcată cu o linie groasă, unde am găsit mutațiile fondatoare în genele KCNQ1 și KCNH2, descrise în familia 1-4.
deoarece LQTS este o boală autozomală dominantă, ne-am așteptat să observăm pacienți purtători de mutații predominant heterozigoți. Dar în studiul nostru, am identificat în principal pacienți cu mutații recesive (12-14). În mod evident, acești pacienți devin mai întâi simptomatici și, au fost referiți predominant la centrul cardiac Prince Sultan, aducându-i sub atenția noastră. Cu toate acestea, constatările din investigațiile noastre implică faptul că LQTS recesive ar putea avea fenotipuri clinice fatale la copii și nu sunt neobișnuite în Arabia Saudită (12-14). Deoarece purtătorii de mutații heterozigote sunt, de asemenea, susceptibili de a dezvolta aritmie și complicațiile acesteia, ar trebui luată o inițiativă concertată pentru a aduce medicii generali locali, cardiologii și geneticienii clinici într-o platformă comună pentru a identifica persoanele cu risc. Mai mult, în acest moment, nu avem nici o informație genetică pentru alte tulburări de aritmie familială, de exemplu, CPVT, SQTS, BrS, AF etc. Centrele cardiogenetice specializate ar trebui să ia inițiativa de a căuta defectele genetice, mutațiile și de a efectua studii genotip-fenotip în toate formele de aritmii ereditare. De asemenea, trebuie luat în considerare faptul că nu toate aritmiile genetice ar avea antecedente familiale, deoarece în multe cazuri mutația cauzală a aritmiei este de origine de novo, adică probandul este primul pacient din familia respectivă cu mutația și el/ea este sursa de transmitere a mutației în generațiile din aval (110, 111). Datorită ratei foarte ridicate a căsătoriilor consanguine din Arabia Saudită, ne așteptăm ca mult mai multe mutații fondatoare să exercite un rol crucial în aritmiile congenitale din această țară. Identificarea acestor mutații fondatoare ar trebui să fie prima și cea mai importantă sarcină a noastră, care ne-ar facilita dezvoltarea unei consiliere genetică eficientă premaritală și, de asemenea, pre-simptomatică în această țară. Mutațiile genelor cauzale LQTS ar putea conferi variabilitate în penetranța clinică, într-o extremă, unii purtători ar putea fi probabil complet sănătoși, dar unii purtători ar putea avea prima lor manifestare a bolii ca sincopă sau moarte subită. Moartea subită a unui copil mic sau a unui adult reprezintă o mare povară psihologică și emoțională asupra familiei, screening-ul transportatorilor este esențial, deoarece există medicamente simple, de exemplu, blocante de blocante (de asemenea, modificarea comportamentului), care ar putea preveni foarte eficient purtătorii de consecințele fatale ale aritmiei și SCDs. Persoanele cu mutații homozigote în genele cauzale LQTS ar putea avea morbiditate severă și o rată extrem de mare de mortalitate, inclusiv Brady-tahiaritmii fetale și, în multe cazuri, avorturi spontane, avorturi spontane la mamele însărcinate (12-14).
nu s-au efectuat prea multe cercetări în Arabia Saudită cu privire la prevalența diferitelor SNP-uri în genele legate de aritmie și, de asemenea, în genele care le reglează. Splawski și colab. (112) a descris o variantă comună S1103Y în gena SCN5A asociată cu aritmia la afro-americani. Alela variantă numită Y1103 este responsabilă pentru accelerarea activării canalului, sporind astfel probabilitatea aritmiilor cardiace la persoanele de origine africană (52, 112). K393N este o variantă a genei KCNQ1 raportată la pacienții cu LQTS1 din SUA, dar, în populația arabă, am detectat această variantă la 2% dintre indivizi (date nepublicate). Dacă varianta K393N în gena KCNQ1 la arabi este comparabilă cu varianta S1103y (SCN5A) la afro-americani sau varianta D85N (KCNE1) la populația japoneză ar putea merita, de asemenea, investigații (59).
Declarație privind conflictul de interese
autorii declară că cercetarea a fost realizată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
mulțumiri
recunoștința noastră sinceră Prof.Arthur Wilde, Prof. Connie Bezzina, și editorul revistei “Heart” pentru permisiunea lor de a folosi Figura 1 în acest manuscris din Ref. (113).
1. Bhuiyan ZA. Spectrul clinic și Genetic al sindroamelor ereditare de aritmie cardiacă. Amsterdam: Universitatea Din Amsterdam (2009).
2. Priori SG, Aliot E, bl Oktstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, și colab. Grupul operativ privind moartea subită cardiacă, Societatea Europeană de Cardiologie. Europace (2002) 4:3-18. doi: 10.1053 / eupc.2001.0214
CrossRef Text Complet
3. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL și colab. Mutații SCN5A asociate cu o aritmie cardiacă moștenită, sindrom QT lung. Celulă (1995) 80:805-11. doi:10.1016/0092-8674(95)90359-3
Pubmed rezumat / Pubmed text complet / CrossRef text complet
4. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. O bază moleculară pentru aritmia cardiacă: mutațiile HERG provoacă sindromul QT lung. Celulă (1995) 80:795-803. doi: 10.1016/0092-8674(95)90358-5
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
5. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ și colab. Clonarea pozițională a unei noi gene a canalului de potasiu: mutațiile KVLQT1 provoacă aritmii cardiace. Nat Genet (1996) 12:17-23. doi: 10.1038 / ng0196-17
Pubmed rezumat / Pubmed text complet / CrossRef text complet
6. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutațiile genei hminK provoacă sindromul QT lung și suprimă funcția IKs. Nat Genet (1997) 17:338-40. doi: 10.1038 / ng1197-338
Pubmed rezumat / Pubmed text complet / CrossRef text complet
7. Sanguinetti MC, Jiang C, Curran ME, Keating MT. O legătură mecanică între o aritmie cardiacă moștenită și una dobândită: HERG codifică canalul de potasiu IKr. Celulă (1995) 81:299-307. doi:10.1016/0092-8674(95)90340-2
Pubmed rezumat / Pubmed text complet / CrossRef text complet
8. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, și colab. O nouă mutație a genei canalului de potasiu KVLQT1 provoacă sindromul cardioauditor Jervell și Lange-Nielsen. Nat Genet (1997) 15:186-9. doi: 10.1038 / ng0297-186
Pubmed rezumat | Pubmed text complet | CrossRef text complet
9. Kucera JP, Rohr s, Rudy Y. localizarea canalelor de sodiu în discurile intercalate modulează conducerea cardiacă. Circ Res (2002) 91:1176-82. doi: 10.1161 / 01.RES. 0000046237.54156. 0 a
Pubmed rezumat / Pubmed text complet / CrossRef text complet
10. Oxford EM, Musa H, Maass K, Coombs W, TAFFET SM, Delmar M. Connexin43 remodelare cauzată de inhibarea expresiei plakophilin-2 în celulele cardiace. Circ Res (2007) 101:703-11. doi: 10.1161 / CIRCRESAHA.107.154252
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
11. Rohr S. crosstalk Molecular între joncțiunile mecanice și electrice la discul intercalat. Circ Res (2007) 101:637-9. doi: 10.1161 / CIRCRESAHA.107.161901
CrossRef Text Complet
12. Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS și colab. Pierderea fetală intrauterină recurentă datorată absenței apropiate a HERG: caracterizarea clinică și funcțională a unei mutații homozigote nonsens HERG Q1070X. Ritmul Cardiac (2008) 5:553-61. doi: 10.1016 / j.hrthm.2008.01.020
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
13. Bhuiyan ZA, Momenah TS, Amin TS, al-Khadra AS, Alders M, Wilde Aam și colab. O mutație Intronică care duce la omiterea incompletă a exon-2 în KCNQ1 salvează auzul în sindromul Jervell și Lange-Nielsen. Prog Biophys Mol Biol (2008) 98:319-27. doi: 10.1016 / j. pbiomolbio.2008.10.004
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
14. Bhuiyan ZA, Al-Shahrani S, Al-Khadra AS, Al-Ghamdi s, Al-Kalaf K, Mannens MMAM, și colab. Analiza clinică și genetică a sindromului QT lung la copiii din șase familii din Arabia Saudită: sunt diferiți? Pediatr Cardiol (2009) 30:490-501. doi: 10.1007 / s00246-008-9377-y
Pubmed rezumat / Pubmed text complet / CrossRef text complet
15. Shinwari ZM, al-Hazzani A, Dzimiri N, Tulbah S, Mallawi Y, Al-Fayyadh M, și colab. Identificarea unei noi mutații KCNQ1 într-o mare familie Saudită cu sindrom QT lung: consecințe clinice și implicații preventive. Clin Genet (2013) 83:370-4. doi: 10.1111 / j. 1399-0004.2012. 01914.x
Pubmed rezumat / Pubmed text complet / CrossRef text complet
16. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana a, Bosi G, și colab. Prevalența sindromului congenital de QT lung. Circulație (2009) 120:1761-7. doi: 10.1161 / CIRCULATIONAHA.109.863209
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
17. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Criterii de Diagnostic pentru sindromul QT lung. O actualizare. Circulație (1993) 88:782-4. doi: 10.1161 / 01.CIR.88.2.782
CrossRef Text Complet
18. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, și colab. Rezumat executiv: declarație de consens HRS/Ehra/APHRS privind diagnosticul și gestionarea pacienților cu sindroame de aritmie primară moștenită. Europace (2013) 15:1389-406. doi: 10.1093/europace / eut272
CrossRef text complet
19. Liu JF, Jons C, Moss AJ, McNitt S, Peterson Dr, Qi M, și colab. Registrul Sindromului. Factori de risc pentru sincopă recurentă și evenimente ulterioare fatale sau aproape fatale la copii și adolescenți cu sindrom QT lung. J Am Coll Cardiol (2011) 57:941-50. doi: 10.1016 / j.jacc.2010.10.025
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
20. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL și colab. Spectrul mutațiilor în genele sindromului QT lung. KVLQT1, HERG, SCN5A, KCNE1 și KCNE2. Circulație (2000) 102:1178-85. doi: 10.1161 / 01.CIR.102.10.1178
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
21. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Mutații compuse: o cauză comună a sindromului QT lung sever. Circulație (2004) 109:1834-41. doi: 10.1161 / 01.CIR.0000125524.34234.13
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
22. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, și colab. Risc de evenimente cardiace care pun viața în pericol în rândul pacienților cu sindrom QT lung și mutații multiple. Ritmul Cardiac (2013) 10:378-82. doi: 10.1016 / j.hrthm.2012.11.006
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
23. Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, și colab. Testarea genetică în sindromul QT lung: dezvoltarea și validarea unei abordări eficiente a genotipării în practica clinică. JAMA (2005) 294:2975-80. doi: 10.1001 / jama.294.23.2975
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
24. Roden DM. Practica clinică. Sindromul QT lung. N Engl J Med (2008) 358:169-76. doi: 10.1056 / NEJMcp0706513
CrossRef text complet
25. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C și colab. Corelația genotip-fenotip în sindromul QT lung: declanșatori specifici genei pentru aritmii care pun viața în pericol. Circulație (2001) 103:89-95. doi: 10.1161 / 01.CIR.103.1.89
CrossRef Text Complet
26. Ackerman MJ, Tester DJ, Porter CJ. Înot, un declanșator aritmogen specific genei pentru sindromul QT lung moștenit. Mayo Clin Proc (1999) 74:1088-94. doi:10.4065/74.11.1088
Pubmed rezumat / Pubmed text complet / CrossRef text complet
27. Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, și colab. Testarea genetică pentru sindromul QT lung: distingerea mutațiilor patogene de variantele benigne. Circulație (2009) 120:1752-60. doi: 10.1161 / CIRCULATIONAHA.109.863076
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
28. Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, și colab. Aspecte clinice ale sindromului QT lung de tip 1 în funcție de locație, tip de codificare și funcția biofizică a mutațiilor care implică gena KCNQ1. Circulație (2007) 115:2481-9. doi: 10.1161 / CIRCULATIONAHA.106.665406
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
29. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, și colab. Variantele din regiunea netradusă 3′ a canalului de potasiu Kv7.1 codificat KCNQ1 modifică severitatea bolii la pacienții cu sindrom QT lung de tip 1 într-o manieră specifică alelei. Eur Inima J (2012) 33:714-23. doi: 10.1093 / eurheartj / ehr473
Pubmed rezumat / Pubmed text complet / CrossRef text complet
30. Barsheshet A, Goldenberg I, o-Uchi J, Moss AJ, Jons C, Shimizu W, și colab. Mutații în buclele citoplasmatice ale canalului KCNQ1 și riscul de evenimente care pun viața în pericol: implicații pentru răspunsul specific mutației la terapia cu blocante ale receptorilor de calciu în sindromul QT lung de tip 1. Circulație (2012) 125:1988-96. doi: 10.1161 / CIRCULATIONAHA.111.048041
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
31. Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, și colab. Sindromul Jervell și Lange-Nielsen: Istorie Naturală, bază moleculară și rezultat clinic. Circulație (2006) 113:783-90. doi: 10.1161 / CIRCULATIONAHA.105.592899
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
32. Bhuiyan ZA, Wilde AA. IKs în inimă și auz, urechea poate face cu mai puțin decât inima. Circ Cardiovasc Genet (2013) 6:141-3. doi: 10.1161 / CIRCGENETICS.113.000143
CrossRef Text Complet
33. Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Mecanisme fiziopatologice ale mutațiilor dominante și recesive ale canalului KVLQT1 k + găsite în aritmiile cardiace moștenite. Hum Mol Genet (1997) 6:1943-9. doi: 10.1093 / hmg / 6.11.1943
CrossRef Text Complet
34. Wilde AA, Jongbloed RJ, Doevendans PA, d Untren DR, Hauer RN, van Langen IM, și colab. Stimulii auditivi ca declanșator al evenimentelor aritmice diferențiază pacienții legați de HERG (LQTS2) de pacienții legați de KVLQT1 (LQTS1). J Am Coll Cardiol (1999) 33:327-32. doi: 10.1016 / S0735-1097(98)00578-6
CrossRef text complet
35. Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, și colab. Risc crescut de evenimente aritmice în sindromul QT lung cu mutații în regiunea porilor canalului de potasiu al genei umane legate de eter-a-go-go. Circulație (2002) 105:794-9. doi: 10.1161 / hc0702. 105124
Pubmed rezumat | Pubmed text complet | CrossRef text complet
36. Lupoglazoff JM, Denjoy I, ticălos E, Fressart V, Simon F, Bozio A, și colab. Sindromul QT lung la nou-născuți: tulburări de conducere asociate cu mutații HERG și bradicardie sinusală cu mutații KCNQ1. J Am Coll Cardiol (2004) 43:826-30. doi: 10.1016 / j.jacc.2003.09.049
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
37. Chevalier P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ și colab. Torsada vârfurilor complică blocul atrioventricular: dovezi pentru o predispoziție genetică. Ritmul Cardiac (2007) 4:170-4. doi: 10.1016 / j.hrthm.2006.10.004
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
38. Hoorntje T, Alders M, van Tintelen P, van der Lip K, Sreeram N, van der Wal A, și colab. Trunchierea prematură homozigotă a proteinei HERG: human HERG knockout. Circulație (1999) 100:1264-7. doi: 10.1161 / 01.CIR.100.12.1264
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
39. Piippo K, Laitinen P, Swan H, Toivonen L, Viitasalo M, Pasternack M și colab. Homozigozitatea pentru o mutație a canalului de potasiu HERG determină o formă severă de sindrom QT lung: identificarea unei mutații fondatoare aparente la finlandezi. J Am Coll Cardiol (2000) 35:1919-25. doi: 10.1016 / S0735-1097(00)00636-7
Pubmed rezumat / Pubmed text complet / CrossRef text complet
40. Johnson WH Jr, Yang P, Yang T, Lau YR, Mostella BA, Wolff DJ, și colab. Caracterizarea clinică, genetică și biofizică a unei mutații HERG homozigote care provoacă sindrom QT lung neonatal sever. Pediatru Res (2003) 53:744-8. doi: 10.1203 / 01.PDR.0000059750.17002.B6
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
41. Ackerman MJ, Tester DJ, Jones GS, va ML, vizuină CR, Curran mine. Diferențe etnice în variantele canalului cardiac de potasiu: implicații pentru susceptibilitatea genetică la moarte subită cardiacă și testarea genetică pentru sindromul QT lung congenital. Mayo Clin Proc (2003) 78:1479-87. doi:10.4065/78.12.1479
Pubmed rezumat / Pubmed text complet / CrossRef text complet
42. Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, de Ferrari GM și colab. KCNH2-K897T este un modificator genetic al sindromului congenital latent de QT lung. Circulație (2005) 112:1251-8. doi: 10.1161 / CIRCULATIONAHA.105.549071
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
43. Nof E, Cordeiro JM, P Otrivrez GJ, Scornik FS, Calloe K, dragoste B, și colab. Un polimorfism nucleotidic unic comun poate exacerba sindromul QT lung de tip 2 care duce la moartea subită a sugarului. Circ Cardiovasc Genet (2010) 3:199-206. doi: 10.1161 / CIRCGENETICS.109.898569
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
44. Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, și colab. O singură mutație a canalului Na ( + ) care provoacă atât sindroame QT lung, cât și sindroame Brugada. Circ Res (1999) 85:1206-13. doi: 10.1161 / 01.RES.85.12.1206
Pubmed rezumat / Pubmed text complet / CrossRef text complet
45. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, și colab. Noua mutație SCN5A care duce fie la defect izolat de conducere cardiacă, fie la sindromul Brugada într-o mare familie franceză. Circulație (2001) 104:3081-6. doi: 10.1161 / hc5001. 100834
Pubmed rezumat | Pubmed text complet | CrossRef text complet
46. Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, și colab. Mutația E1784K în SCN5A este asociată cu fenotipul clinic mixt al sindromului QT lung de tip 3. J Clin Invest (2008) 118:2219-29. doi: 10.1172 / JCI34057
Pubmed rezumat / Pubmed text complet / CrossRef text complet
47. Keller DI, Acharfi S, Delacr Inktaz e, Benammar N, Rotter M, Pfammatter JP și colab. O nouă mutație în SCN5A, delQKP 1507-1509, provocând sindromul QT lung: rolul reziduului Q1507 în inactivarea canalului de sodiu. J Mol Cell Cardiol (2003) 35:1513-21. doi: 10.1016 / j. yjmcc.2003.08.007
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
48. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, și colab. Stratificarea riscului în sindromul QT lung. N Engl J Med (2003) 348:1866-74. doi: 10.1056 / NEJMoa022147
Pubmed rezumat / Pubmed text complet / CrossRef text complet
49. Lupoglazoff JM, Cheav T, Baroudi G, Berthet M, Denjoy I, Cauchemez B și colab. Mutație homozigotă SCN5A în sindromul QT lung cu bloc atrioventricular funcțional doi la unu. Circ Res (2001) 89:E16–21. doi: 10.1161 / hh1401.095087
CrossRef text complet
50. Shi R, Zhang Y, Yang c, Huang c, Zhou X, Qiang H, și colab. Mutația cardiacă a canalului de sodiu delQKP 1507-1509 este asociată cu spectrul fenotipic în expansiune al LQT3, tulburarea de conducere, cardiomiopatia dilatativă și incidența ridicată a morții subite a tinerilor. Europace (2008) 10:1329-35. doi: 10.1093 / europace / eun202
Pubmed rezumat / Pubmed text complet / CrossRef text complet
51. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartscheer A, Eckardt L, Paul M și colab. O mutație de deleție heterozigotă în gena canalului de sodiu cardiac SCN5A cu caracteristici de pierdere și câștig de funcție se manifestă ca boală de conducere izolată, fără semne de Brugada sau sindrom QT lung. PLoS Unul (2013) 8:e67963. doi: 10.1371/jurnal.pone.0067963
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
52. Plant LD, Bowers PN, Liu Q, Morgan T, Zhang t, Stat MW, și colab. O variantă comună a canalului cardiac de sodiu asociată cu moartea subită a sugarului la afro-americani, SCN5A S1103y. J Clin Invest (2006) 116:430-5. doi: 10.1172 / JCI25618
Pubmed rezumat / Pubmed text complet / CrossRef text complet
53. Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH și colab. Mutația Ankyrin-B provoacă aritmie cardiacă de tip 4 cu QT lung și moarte subită cardiacă. Natură (2003) 421:634-9. doi: 10.1038 / nature01335
Pubmed rezumat / Pubmed text complet / CrossRef text complet
54. Schott JJ, Charpentier F, Peltier s, Foley P, Drouin E, Bouhour JB și colab. Cartografierea unei gene pentru sindromul QT lung la cromozomul 4q25-27. Am J Hum Genet (1995) 57:1114-22.
Rezumat Pubmed / Text Integral Pubmed
55. Barhanin J, Lesage F, Guillemare e, Fink M, Lazdunski M, Romey G. K(V)lqt1 și LSK (nurca) proteinele se asociază pentru a forma curentul cardiac de potasiu i(Ks). Natură (1996) 384:78-80. doi: 10.1038 / 384078a0
Pubmed rezumat / Pubmed text complet / CrossRef text complet
56. Sanguinetti MC, Curran ME, Zou a, Shen J, Spector PS, Atkinson DL și colab. Coasamblarea proteinelor K (V)LQT1 și minK(IsK) pentru a forma canalul de potasiu cardiac i (Ks). Natură (1996) 384:80-3. doi: 10.1038 | 384080a0
Pubmed rezumat / Pubmed text complet / CrossRef text complet
57. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y și colab. Mutațiile KCNE1 provoacă sindromul Jervell și Lange-Nielsen. Nat Genet (1997) 17:267-8. doi: 10.1038 / ng1197-267
CrossRef text complet
58. Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutația genei pentru IKs asociată atât cu formele jervell și Lange-Nielsen, cât și cu Romano-Ward ale sindromului QT lung. Circulație (1998) 97:142-6. doi: 10.1186/1471-2350-9-24
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
59. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ și colab. D85n, un polimorfism KCNE1, este o variantă genetică cauzatoare de boli în sindromul QT lung. J Am Coll Cardiol (2009) 54:812-9. doi: 10.1016 / j.jacc.2009.06.005
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
60. Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, și colab. Variațiile genetice ale KCNQ1, KCNH2, SCN5A, KCNE1 și KCNE2 la pacienții cu sindrom QT lung indus de medicamente. Mol Med (2004) 82:182-8. doi: 10.1007 / s00109-003-0522-z
Pubmed rezumat / Pubmed text complet / CrossRef text complet
61. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW și colab. MiRP1 formează canale de potasiu IKr cu HERG și este asociat cu aritmie cardiacă. Celulă (1999) 97:175-87. doi: 10.1016 / S0092-8674(00)80728-X
Pubmed rezumat | Pubmed text complet / CrossRef text complet
62. Gordon e, Panaghie G, Deng L, Bee KJ, Roepke TK, Krogh-Madsen T, și colab. O mutație KCNE2 la un pacient cu aritmie cardiacă indusă de stimuli auditivi și dezechilibru electrolitic seric. Cardiovasc Res (2008) 77:98-106. doi: 10.1093 / cvr / cvm030
Pubmed rezumat / Pubmed text complet / CrossRef text complet
63. Ipsos NM, Tawil R, Tristani-Firouzi M, poate s, Bendahhou s, Tsunoda a, și colab. Mutațiile din Kir2.1 determină fenotipurile electrice de dezvoltare și episodice ale sindromului Andersen. Celulă (2001) 105:511-9. doi: 10.1016 / S0092-8674(01)00342-7
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
64. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Structura primară și expresia funcțională a unui canal de potasiu redresor interior al mouse-ului. Natură (1993) 362:127-33. doi: 10.1038 / 362127a0
Pubmed rezumat / Pubmed text complet / CrossRef text complet
65. Raab-Graham KF, Radeke CM, Vandenberg CA. Clonarea moleculară și exprimarea unui canal de potasiu redresor interior al inimii umane. Neuroreport (1994) 5:2501-5. doi: 10.1097/00001756-199412000-00024
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
66. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R și colab. Ca (V)1.2 disfuncția canalului de calciu determină o tulburare multisistemică, inclusiv aritmie și autism. Celulă (2004) 119:19-31. doi: 10.1016 / j.celulă.2004.09.011
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
67. Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, și colab. Tulburare severă de aritmie cauzată de mutații cardiace ale canalelor de calciu de tip L. Proc Natl Acad Sci U S A (2005) 102:8089-96. doi: 10.1073 / pnas.0502506102
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
68. Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L, și colab. QT lung, sindactilie, contracturi articulare, accident vascular cerebral și mutație CACNA1C nouă: extinderea spectrului sindromului Timothy. Am J Med Genet A (2012) 158A:182-7. doi: 10.1002 / ajmg.a.34355
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
69. Dufendach KA, Giudicessi JR, Boczek NJ, Ackerman MJ. Mozaicismul matern confundă diagnosticul neonatal al sindromului Timothy de tip 1. Pediatrie (2013) 131:e1991–5. doi: 10.1542 / peds.2012-2941
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
70. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, și colab. Mutant caveolin-3 induce curent persistent de sodiu târziu și este asociat cu sindromul QT lung. Circulație (2006) 114:2104–12. doi: 10.1161 / CIRCULATIONAHA.106.635268
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
71. Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC și colab. Mecanism nou pentru sindromul morții subite a sugarului: curent persistent de sodiu târziu secundar mutațiilor din caveolin-3. Ritmul Cardiac (2007) 4:161-6. doi: 10.1016 / j.hrthm.2006.11.030
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
72. Engelman JA, Zhang X, Galbiati F, Volonte D, Sotgia F, Pestell RG, și colab. Genetica moleculară a familiei genei caveolin: implicații pentru cancerele umane, diabetul, boala Alzheimer și distrofia musculară. Am J Hum Genet (1998) 63:1578-87. doi:10.1086/302172
CrossRef text complet
73. Balijepalli RC, Foell JD, Sala DD, iad JW, Kamp TJ. Localizarea canalelor cardiace ca(2+) de tip l la un complex de semnalizare macromolecular caveolar este necesară pentru reglarea beta(2)-adrenergică. Proc Natl Acad Sci U S A (2006) 103:7500-5. doi: 10.1073 / pnas.0503465103
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
74. Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B și colab. SCN4B-codificat canal de sodiu beta4 subunitate în sindromul QT lung congenital. Circulație (2007) 116:134-42. doi: 10.1161 / CIRCULATIONAHA.106.659086
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
75. Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutația unei proteine de ancorare a-kinazei provoacă sindromul QT lung. Proc Natl Acad Sci U S A (2007) 104:20990-5. doi: 10.1073 / pnas.0710527105
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
76. Wu G, ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, și colab. Mutația alfa-1-sintrofină și sindromul QT lung: o boală a perturbării canalului de sodiu. Circ Arrhythm Electrofiziol (2008) 1:193-201. doi: 10.1161 / CIRCEP.108.769224
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
77. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, și colab. Identificarea unui Kir3.4 mutație în sindromul QT lung congenital. Am J Hum Genet (2010) 86:872-80. doi: 10.1016 / j.ajhg.2010.04.017
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
78. Roden DM. Mecanisme și gestionarea proaritmiei. Am J Cardiol (1998) 82:49I–57I. doi:10.1016/S0002-9149(98)00472-X
CrossRef text complet
79. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. O bază structurală pentru sindromul QT lung indus de medicamente. Proc Natl Acad Sci U S A (2000) 97:12329-33. doi: 10.1073 / pnas.210244497
CrossRef Text Complet
80. Kannankeril PJ, Roden DM. Induse de droguri QT lung și torsada vârfurilor: progrese recente. Curr Opin Cardiol (2007) 22:39-43. doi: 10.1097 / HCO.0b013e32801129eb
Pubmed rezumat / Pubmed text complet / CrossRef text complet
81. Veerman CC, Verkerk AO, Blom MT, Klemens CA, Langendijk PN, van Ginneken AC și colab. Lent întârziat blocajul curent redresor potasiu contribuie important la induse de droguri sindromul QT lung. Circ Arrhythm Electrofiziol (2013) 6:1002-9. doi: 10.1161 / CIRCEP.113.000239
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
82. Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ și colab. Un polimorfism comun asociat cu aritmia cardiacă indusă de antibiotice. Proc Natl Acad Sci U S A (2000) 97:10613-8. doi: 10.1073 / pnas.180223197
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
83. K inktibb s, Crawford DC, păcătos MF, Behr er, kannankeril PJ, Wilde AA, și colab. Un sondaj genic candidat mare identifică polimorfismul KCNE1 D85N ca un posibil modulator al torsadelor de vârfuri induse de medicamente. Circ Cardiovasc Genet (2012) 5:91-9. doi: 10.1161 / CIRCGENETICS.111.960930
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
84. K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase S, și colab. Sindromul QT lung indus de anticorpi anti-KCNH2: formă nouă dobândită de sindrom QT lung. J Am Coll Cardiol (2007) 50:1808-9. doi: 10.1016 / j.jacc.2007.07.037
CrossRef Text Complet
85. Moss AJ, Zareba W, Benhorin J, Locati EH, Sala WJ, Robinson JL, și colab. ECG tipare de undă T în forme distincte genetic ale sindromului QT lung ereditar. Circulație (1995) 92:2929-34. doi: 10.1161 / 01.CIR.92.10.2929
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
86. Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC și colab. Spectrul modelelor de undă ST-T și parametrii de repolarizare în sindromul QT lung congenital: constatările ECG identifică genotipurile. Circulație (2000) 102:2849-55. doi: 10.1161 / 01.CIR.102.23.2849
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
87. Tan HL, Bardai A, Shimizu W, Moss AJ, Schulze-Bahr E, Noda T, și colab. Debutul specific al genotipului aritmiilor în sindromul congenital de QT lung: posibile implicații ale terapiei. Circulație (2006) 114:2096-103. doi: 10.1161 / CIRCULATIONAHA.106.642694
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
88. Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH și colab. Diferențe legate de vârstă și sex în manifestările clinice la pacienții cu sindrom congenital de QT lung: constatări din registrul internațional LQTS. Circulație (1998) 97:2237-44. doi: 10.1161 / 01.CIR.97.22.2237
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
89. Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG și colab. Influența genotipului asupra cursului clinic al sindromului QT lung. Grupul Internațional de cercetare a Registrului sindromului QT lung. N Engl J Med (1998) 339:960-5. doi: 10.1056 / NEJM199810013391404
Pubmed rezumat / Pubmed text complet / CrossRef text complet
90. Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson Dr, McNitt S, și colab. Sindromul QT lung după vârsta de 40 de ani. Circulație (2008) 117:2192-201. doi: 10.1161 / CIRCULATIONAHA.107.729368
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
91. Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, și colab. Registrul Sindromului. Efectele modulatoare ale vârstei și sexului asupra evoluției clinice a sindromului QT lung după genotip. J Am Coll Cardiol (2003) 42:103-9. doi: 10.1016 / S0735-1097(03)00554-0
Pubmed rezumat / Pubmed text complet / CrossRef text complet
92. Seth R, Moss AJ, McNitt S, Zareba W, Andrews ML, Qi M și colab. Sindromul QT lung și sarcina. J Am Coll Cardiol (2007) 49:1092-8. doi: 10.1016 / j.jacc.2006.09.054
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
93. Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, și colab. O legătură moleculară între sindromul morții subite a sugarului și sindromul QT lung. N Engl J Med (2000) 343:262-7. doi: 10.1056 / NEJM200007273430405
CrossRef text complet
94. Schwartz PJ, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini a, și colab. Diagnosticul Molecular la un copil cu sindrom de moarte subită a sugarului. Lancet (2001) 358:1342-3. doi: 10.1016 / S0140-6736(01)06450-9
Pubmed rezumat / Pubmed text complet / CrossRef text complet
95. Christiansen M, T N, Larsen LA, Andersen PS, Simonsen H, Oyen N, și colab. Mutații în canalul HERG K + – ion: o legătură Nouă între sindromul QT lung și sindromul morții subite a sugarului. Am J Cardiol (2005) 95:433-4. doi: 10.1016/j.amjcard.2004.09.054
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
96. Tester DJ, Ackerman MJ. Postmortem sindromul QT lung testarea genetică pentru moarte subită inexplicabilă la tineri. J Am Coll Cardiol (2007) 49:240-6. doi: 10.1016 / j.jacc.2006.10.010
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
97. Hofman N, Tan HL, Clur SA, Alders M, van Langen IM, Wilde AA. Contribuția bolilor cardiace moștenite la moartea subită cardiacă în copilărie. Pediatrie (2007) 120:e967–73. doi: 10.1542 / peds.2006-3751
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
98. Wedekind H, Smits JP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, și colab. Mutația de novo a genei SCN5A asociată cu debutul precoce al morții subite a sugarului. Circulație (2001) 104:1158-64. doi: 10.1161 / hc3501. 095361
CrossRef text complet
99. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M și colab. Disfuncție cardiacă a canalului de sodiu în sindromul morții subite a sugarului. Circulație (2007) 115:368-76. doi: 10.1161 / CIRCULATIONAHA.106.646513
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
100. Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, Londra B, Makielski JC și colab. Caracterizarea moleculară și funcțională a mutațiilor genei asemănătoare glicerolului-3-fosfat dehidrogenazei 1 (GPD1-L) în sindromul morții subite a sugarului. Circulație (2007) 116:2253-9. doi: 10.1161 / CIRCULATIONAHA.107.704627
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
101. Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, și colab. Sindromul morții subite a sugarului-mutații asociate în subunitățile beta ale canalului de sodiu. Ritmul Cardiac (2010) 7:771-8. doi: 10.1016 / j.hrthm.2010.01.032
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
102. Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, și colab. Pierderea fetală recurentă în trimestrul al treilea și mozaicismul matern pentru sindromul QT lung. Circulație (2004) 109:3029-34. doi: 10.1161 / 01.CIR.0000130666.81539.9 E
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
103. Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF și colab. Nu toate beta-blocantele sunt egale în gestionarea tipurilor de sindrom QT lung 1 și 2: recurențe mai mari ale evenimentelor sub Metoprolol. J Am Coll Cardiol (2012) 60:2092-6. doi: 10.1016 / j.jacc.2012.07.046
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
104. Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C și colab. Denervarea simpatică cardiacă stângă în managementul pacienților cu risc crescut afectați de sindromul QT lung. Circulație (2004) 109:1826-33. doi: 10.1161 / 01.CIR.0000125523.14403.1 E
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
105. Singh B, Al Shahwan SA, Habbab MA, Al Deeb SM, Biary N. sindromul QT lung idiopatic: punând întrebarea corectă. Lanceta (1993) 341:741. doi:10.1016/0140-6736(93)90501-7
CrossRef text complet
106. Gorgels AP, al Fadley F, Zaman L, Kantoch MJ, al Halees Z. sindromul QT lung cu conducere atrioventriculară afectată: o variantă malignă la sugari. J Cardiovasc Electrofiziol (1998) 9:1225-32. doi: 10.1111 / j. 1540-8167.1998.tb00096.x
Pubmed rezumat / Pubmed text complet / CrossRef text complet
107. Kantoch MJ, Qurashi MM, Bulbul ZR, Gorgels AP. Un nou-născut cu o boală cardiacă congenitală complexă, bloc atrioventricular și tahicardie ventriculară torsada vârfurilor. Stimularea Clinului Electrofiziol (1998) 21:2664-7. doi: 10.1111 / j. 1540-8159.1998.tb00043.X
CrossRef text complet
108. El-Hazmi MA, al-Swailem AR, Warsy AS, Al-Swailem AM, Sulaimani R, al-Meshari AA. Consangvinitate în rândul populației din Arabia Saudită. J Med Genet (1995) 32:623-6. doi: 10.1136/jmg.32.8.623
CrossRef Text Complet
109. El Mouzan MI, al Salloum AA, Al Herbish AS, Qurachi MM, Al Omar AA. Consanguinitatea și tulburările genetice majore la copiii Saudiți: un studiu transversal bazat pe comunitate. Ann Saudi Med (2008) 28:169-73. doi:10.4103/0256-4947.51726
Pubmed rezumat / Pubmed text complet / CrossRef text complet
110. Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van Tintelen JP și colab. Receptorul ryanodinei/canalul de eliberare a calciului codificat RYR2 la pacienții diagnosticați anterior fie cu tahicardie ventriculară polimorfă catecolaminergică, fie cu sindrom QT lung indus de genotip negativ, indus de efort: o analiză mutațională cuprinzătoare a cadrului de citire deschis. J Am Coll Cardiol (2009) 54:2065-74. doi: 10.1016 / j.jacc.2009.08.022
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
111. Al-Aama JY, al-Ghamdi S, Bdier AY, Wilde AA, Bhuiyan ZA. Mutația de novo în gena KCNQ1 cauzală sindromului Jervell și Lange-Nielsen. Clin Genet (2013). doi: 10.1111 / cge.12300
Pubmed Rezumat / Pubmed Text Complet / CrossRef Text Complet
112. Splawski I, Timothy KW, Tateyama M, Clancy CE, Malhotra a, Beggs AH, și colab. Varianta canalului de sodiu SCN5A implicat în riscul de aritmie cardiacă. Știință (2002) 297:1333-6. doi: 10.1126 / știință.1073569
CrossRef Text Complet
113. Wilde AA, Bezzina CR. Genetica aritmiilor cardiace. Inimă (2005) 91:1352-8.