Genetica buzelor despicate și a Palatului
 intro/abstractbuza stângă cu sau fără Palatul despicat este o anomalie congenitală complexă care poate fi izolată sau văzută împreună cu alte malformații. De asemenea, poate face parte din fenotipul unui sindrom genetic. Acest articol servește ca o revizuire a prevalenței buzelor și palatului despicat, a riscurilor de recurență și a riscurilor pentru alte anomalii congenitale. Vor fi explorate sindroamele genetice și expunerile teratogene cunoscute a fi asociate cu fisuri orale. În plus, vor fi discutate testele genetice solicitate în mod obișnuit în cadrul geneticii clinice pediatrice pentru evaluarea pacientului cu buza și Palatul despicat.
intro/abstractbuza stângă cu sau fără Palatul despicat este o anomalie congenitală complexă care poate fi izolată sau văzută împreună cu alte malformații. De asemenea, poate face parte din fenotipul unui sindrom genetic. Acest articol servește ca o revizuire a prevalenței buzelor și palatului despicat, a riscurilor de recurență și a riscurilor pentru alte anomalii congenitale. Vor fi explorate sindroamele genetice și expunerile teratogene cunoscute a fi asociate cu fisuri orale. În plus, vor fi discutate testele genetice solicitate în mod obișnuit în cadrul geneticii clinice pediatrice pentru evaluarea pacientului cu buza și Palatul despicat.
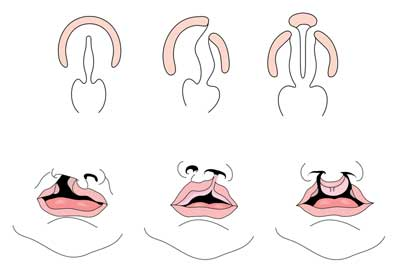
buza Cleft cu sau fără palatoschizis (CL/CP) diferă de un palatoschizis izolat (CP) la nivel embrionar, epidemiologic și genetic. Buza despicată rezultă de obicei din proeminența maxilară și proeminența nazală mediană care nu se contopesc între a cincea și a șasea săptămână de dezvoltare embrionară. Dezvoltarea normală a Palatului rezultă din formarea Palatului primar și a Palatului secundar. Palatul primar se formează în săptămânile șase până la șapte prin dezvoltarea și fuziunea proceselor nazale mediale, nazale laterale și maxilare. Palatul secundar provine din rafturile palatale (care se dezvoltă din procesele maxilare pereche ale primului arc ramificat) devenind orizontal și fuzionând, formând Palatele dure și moi în jurul celei de-a noua săptămâni de dezvoltare embrionară. Rafturile fuzionează, de asemenea, cu Palatul primar și septul nazal. (1)
fisurile orale sunt unul dintre cele mai frecvente defecte congenitale observate în pepiniera neonatală, cu o prevalență generală de 1.6 la mia de nou-născuți la nivel mondial, cu CL/CP Văzut la aproximativ o mie de nașteri și CP Văzut la 0,6 la mia de nașteri. (2) există o frecvență mai mare a CL/CP la indivizii de origine asiatică, africană și nativă americană. CL / CP este, de asemenea, mai frecvent la bărbați. În schimb, nu există o diferență semnificativă în incidența CP între diferite medii etnice, iar CP este mai frecventă la femei. (3) riscurile de recurență în cadrul unei familii depind de faptul dacă fisura este izolată (fără alte constatări clinice prezente) sau văzută ca parte a unui sindrom genetic. Cele mai multe cazuri de fisuri orale sunt izolate (aproximativ 80%). Se crede că fisurile izolate au moștenire multifactorială: se datorează unei combinații de factori multipli, atât genetici, cât și de mediu. Riscul de recurență (Tabelul 1) crește atunci când există mai multe rude afectate. Riscul de recurență crește, de asemenea, cu cât defectul este mai sever.

buza despicată și Palatul pot fi observate cu alte anomalii congenitale. Probabilitatea unei etiologii genetice sau teratogene crește cu atât mai multe anomalii congenitale cu care se prezintă un pacient. Prezența altor probleme, cum ar fi dizabilitatea intelectuală, problemele de comportament, cum ar fi autismul, trăsăturile dismorfice sau alte preocupări medicale, vor face, de asemenea, o tulburare genetică sau o expunere teratogenă mai probabilă. Aproximativ 13% dintre persoanele cu buza despicată vor avea alte preocupări medicale sau anomalii. Numărul crește la 37% cu buza despicată și palatul și la 47% doar cu Palatul despicat.
expunerea prenatală la agenți teratogeni (cum ar fi talidomida, anticonvulsivantele, alcoolul, acidul retinoic și țigările) și bolile materne (cum ar fi diabetul, rubeola și deficitul de folat) s-au dovedit a crește riscul de fisuri orale. Prezența benzilor amniotice crește, de asemenea, riscul de fisuri. Se știe că suplimentarea cu acid folic Periconceptual reduce riscul de fisuri orale.
secvența Pierre Robin este o anomalie Craniofacială caracterizată prin hipoplazie mandibulară sau micrognatie, palat secundar în formă de U și glossoptoză care duce la apnee obstructivă și dificultăți de hrănire. Secvența Pierre Robin poate fi văzută ca parte a sindroamelor genetice (sindromul de ștergere 22q11.2, sindromul Stickler; descris mai jos). (5)
există sute de sindroame genetice asociate cu fisuri orale, inclusiv anomalii citogenetice (aneuploidii, microdeleții) și tulburări cu o singură genă (Mendeliană). Confirmarea unui diagnostic genetic este esențială pentru a determina prognosticul și a stabili un risc de recurență.
Aneuploidiile precum trisomia 13 și 18 au o asociere puternică cu CL/CP. Trisomia 13 (AKA sindromul Patau) este asociată cu trei copii ale cromozomului 13 sau translocații robertsoniene dezechilibrate care implică cromozomul 13. Bebelușii născuți cu această afecțiune mor de obicei în perioada neonatală. Caracteristicile clinice includ buza despicată și Palatul, întârzierea creșterii, malformații severe ale nervilor centrali (inclusiv holoprosencefalie), microcefalie, microptalmie, colobom iris, absența ochilor, urechi malformate, polidactilie, pumnii încleștați, picioarele inferioare basculante, defecte cardiace congenitale și defecte urogenitale. Fisurile de linie mediană (altfel foarte rare) pot fi observate în trisomia 13 din cauza riscului de defecte ale liniei mediane, inclusiv holoprosencefalie. Trisomia 18 (AKA sindromul Edwards) se datorează de obicei a trei copii distincte ale cromozomului 18 și este asociată cu un rezultat postnatal slab. Caracteristicile clinice includ buza despicată și Palatul, dizabilitatea intelectuală, eșecul de a prospera, bolile cardiace congenitale, hipertonia, micrognatia, sternul scurt, urechile malformate, mâinile încleștate, picioarele inferioare ale rocker-ului și unghiile hipoplastice, printre altele. Trisomia 13 și 18 poate fi ușor confirmată sau exclusă prin efectuarea analizei cromozomiale (cariotipare).
sindroamele de Microdeletie implica de obicei stergerea unei parti a unui cromozom. Aceste ștergeri pot fi prea mici pentru a fi detectate prin cariotipare standard și pot necesita detectarea peștilor (hibridizare fluorescentă in situ) sau a tehnologiei microarray. Un cunoscut sindrom de microdeletie asociat cu Palatul despicat este sindromul de ștergere 22q11.2 (aka sindromul Digeorge/Velocardiofacial). Anomaliile palatale, inclusiv incompetența velofaringiană, fisurile submucoase, uvula bifidă și Palatul despicat sunt observate la 69% dintre persoanele cu deleție 22q11.2 și pot face parte din secvența Pierre Robin. Alte descoperiri clinice includ boli cardiace congenitale, pierderea auzului, caracteristici dismorfice, deficit imunitar, hipocalcemie, anomalii renale, probleme de hrănire, anomalii scheletice și tulburări psihiatrice. Aproximativ 10% din cazurile de sindrom de deleție 22q11.2 sunt considerate a fi familiale. Ștergerea se separă într-un mod dominant autosomal.(6) sindromul Wolf-Hirschhorn, care se datorează unei deleții în brațul scurt al cromozomului 4, este, de asemenea, asociat cu fisuri orale (la 25% până la 50% dintre persoanele afectate). Caracteristicile faciale caracteristice (inclusiv glabella proeminentă care duce la “aspectul căștii războinice grecești”), boli cardiace congenitale, dizabilități intelectuale, convulsii, eșecul de a prospera, micrognatie, etichete sau gropi preauriculare și hipodontie pot fi, de asemenea, văzute ca parte a afecțiunii.(7)
tulburările cu o singură genă cu fisuri orale includ sindromul Stickler, sindromul Treacher Collins și sindromul Van der Woude, printre multe altele. Sindromul Stickler este o tulburare de colagen cu moștenire autosomală dominantă și, mai puțin frecvent, autosomală recesivă. Caracteristicile comune includ Palatul despicat (văzut ca parte a secvenței Pierre Robin sau fără micrognatie), pierderea auzului (senzorineural și conductiv), descoperiri scheletice (artrită cu debut precoce, displazie spondiloepifizală), anomalii oculare (miopie ridicată, anomalii vitroase) și trăsături faciale caracteristice (cu subdezvoltare a maxilarului și a podului nazal, retruzie la mijlocul feței). Testarea genetică pentru sindromul Stickler poate fi complexă, deoarece mutații în cel puțin șase gene au fost descrise la persoanele afectate. Aproximativ 90% dintre pacienții cu sindrom Stickler au mutații în gena COL2A1 și au o formă dominantă autosomală a afecțiunii.(8) sindromul Treacher Collins este o afecțiune autozomală dominantă caracterizată prin Palatul despicat cu sau fără buza despicată la 28% dintre persoanele afectate. Alte anomalii includ hipoplazia oaselor zigomatice și a mandibulei, anomalii ale urechii externe, colobomul pleoapei inferioare, pierderea auzului conductiv, absența genelor inferioare, deplasarea părului preauricular pe obraji și stenoza choanală sau atrezia. Diagnosticul sindromului Treacher Collins se bazează pe constatări clinice și radiografice. Au fost descrise mutații în cel puțin trei gene, cu mutații în TCOF1 observate la 78% până la 93% dintre pacienți.(9) sindromul Van Der Woude se caracterizează prin prezența fistulelor congenitale, de obicei bilaterale, paramediene (gropi), sau uneori mici movile cu un tract sinusal care duce dintr-o glandă mucoasă a buzei și a fisurilor orale (inclusiv CL/CP și CP). Van der Woude este o afecțiune dominantă autosomală asociată cu mutații ale genei IRF6 (10). Testarea Condițiilor cu o singură genă sau cu mai multe gene necesită o analiză directă a genei prin secvențiere și/sau analiză de ștergere/duplicare (cum ar fi MLPA).
având în vedere că sindroamele genetice cu buza despicată și Palatul pot fi asociate cu aneuploidii, microdeleții/microduplicații cromozomiale sau tulburări cu o singură genă, testarea genetică poate fi un proces complicat. Un istoric medical amănunțit, un pedigree de trei generații, un istoric al sarcinii și un examen de dismorfologie efectuat de un genetician clinic pot clarifica tabloul clinic și pot permite testarea genetică vizată. Tehnologiile mai noi, inclusiv microarray, vor permite identificarea microdelețiilor mici și a microduplicațiilor ratate anterior prin cariotiparea standard. Din păcate, această tehnică conduce, de asemenea, la identificarea ștergerilor și duplicărilor de semnificație clinică necunoscută, complicând procesul de consiliere genetică. Testarea tulburărilor cu o singură genă sau a tulburărilor mendeliene necesită disponibilitatea clinică a testelor genetice pentru gena dorită. De asemenea, poate fi scump dacă nu este acoperit de asigurarea medicală. Noile tehnologii, cum ar fi secvențierea de generație următoare, secvențierea exomilor sau secvențierea genomului (cunoscute colectiv sub numele de teste genomice) au devenit acum disponibile clinic. Analizând simultan sute până la mii de gene, aceste teste cresc semnificativ puterea și randamentul diagnosticului. În comparație cu alte tehnici, aceste teste pot oferi un răspuns mai rapid și într-un mod mai rentabil. În domeniul cercetării, secvențierea exomului și a genomului a dus la identificarea de noi gene, precum și la extinderea caracteristicilor clinice și a spectrului pentru mutațiile genetice. Ca și în cazul tehnologiei microarray, testele genomice pot detecta sindroame care nu au legătură cu prezentarea pacientului și/sau motivul testării. Având în vedere complexitatea inerentă a testelor genetice, consimțământul informat este necesar.
concluzie
deși buza și Palatul despicat sunt o anomalie izolată în majoritatea cazurilor, există o asociere puternică între fisurile orale și alte anomalii și sindroame genetice. O evaluare genetică de către un genetician clinic și un consilier genetic este esențială pentru îndrumarea anticipativă și pentru a determina riscurile de recurență. Testarea genetică, care necesită consimțământul informat, poate fi coordonată și interpretată în timpul unei evaluări genetice.
Anya Revah, MS, este consilier genetic senior la Divizia de Genetică Medicală la Spitalul de sugari și copii Maimonides din Brooklyn, New York. De asemenea, este membru activ al Centrului Medical Maimonides și al echipei multidisciplinare Kings County Hospital Cleft Lip and Palate. Are un Master în știință în consiliere genetică de la Universitatea Boston din Boston, Massachusetts.
1. Sadler TW. Embriologia medicală a lui Langman. Ediția A Noua. Paginile 390-395.
2. De asemenea, este important sa va asigurati ca veti beneficia de o gama variata de produse si servicii de calitate. Estimări actualizate ale prevalenței Naționale a nașterii pentru defecte congenitale selectate în Statele Unite. 2004-2006. Cercetarea defectelor congenitale( partea a): Teratologie clinică și moleculară 2010;88: 1008-1016.
3. Fraser FC. Genetica buzelor despicate și a Palatului despicat. Am. J. Hum. Genet. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC și colab. Aportul Periconceptual de folat prin supliment și aport alimentar reduce riscul de buză despicată non-sindromică cu sau fără palat despicat. Med Prev 2004; 39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Perspective de dezvoltare și genetice asupra secvenței Pierre Robin. Am. J. Med. Genet. 2013; 163C: 295-305.
6. McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11. 2 sindromul de ștergere. Sept. 23, 1999. . În: Pagon RA, Adam MP, Ardinger HH și colab., editori. GeneReviews . Seattle( WA): Universitatea din Washington, Seattle; 1993-2014. Disponibil de la: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia A, Carey JC, South ST, și colab. Sindromul Wolf-Hirschhorn. Apr. 29, 2002. . În: Pagon RA, Adam MP, Ardinger HH și colab., editori. GeneReviews . Seattle( WA): Universitatea din Washington, Seattle; 1993-2014. Disponibil de la: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, Ala-Kokko L. sindromul Stickler. Jun. 9, 2000. . În: Pagon RA, Adam MP, Ardinger HH și colab., editori. GeneReviews . Seattle( WA): Universitatea din Washington, Seattle; 1993-2014. Disponibil de la: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs EW. Sindromul Treacher Collins. Iulie. 20, 2004. . În: Pagon RA, Adam MP, Ardinger HH și colab., editori. GeneReviews . Seattle (WA): Universitatea din Washington, Seattle; 1993-2014. Disponibil de la: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy S, și colab. Tulburări legate de IRF6. Oct. 30, 2003. . În: Pagon RA, Adam MP, Ardinger HH și colab., editori. GeneReviews . Seattle( WA): Universitatea din Washington, Seattle; 1993-2014. Disponibil de la: http://www.ncbi.nlm.nih.gov/books/NBK1407/.