Mucopolizaharidozele: semne de diagnostic precoce la sugari și copii
Mucopolizaharidozele (MPS) sunt un grup de boli clinic eterogene cauzate de deficiențe ale enzimelor lizozomale necesare pentru descompunerea glicozaminoglicanilor (gag).
MPS se caracterizează prin manifestări clinice progresive și sistemice. În ciuda eterogenității lor biochimice și genetice, diferite tipuri împărtășesc caracteristici clinice cheie în diferite combinații, inclusiv displazie articulară și scheletică cu rigiditate (cu excepția MPS IV unde există laxitate) și durere, trăsături faciale grosiere, opacifierea corneei, hernii inghinale sau abdominale, infecții recurente ale tractului respirator superior, boli ale valvei cardiace, sindromul tunelului carpian și implicare neurologică variabilă. Aceste caracteristici apar de obicei în primele luni de viață pentru formele severe și în copilăria timpurie pentru formele mai atenuate, dar sunt adesea subestimate și, în general, luate în considerare numai atunci când sunt clar evidente.
raritatea acestor tulburări și variabilitatea prezentării clinice conduc frecvent la o întârziere diagnostică; aceasta poate varia de la luni, pentru formele severe, la ani, pentru cele atenuate (vezi Rigoldi și colab. în acest supliment). Cu toate acestea, datorită progresiei rapide și nevoii urgente de intervenție în prezentările severe, chiar și luni de întârziere în diagnostic pot produce rezultate catastrofale pentru sănătatea viitoare a pacientului.
scopul nostru este de a sublinia în această revizuire semnele foarte timpurii ale formelor severe care ar trebui să alerteze pediatrul la prima lor apariție.
la ce semne / simptome ne putem aștepta la copiii cu forme severe de MPS?
simptomele și semnele sugestive pentru diferite forme de MPS sunt raportate în tabelul 1 în funcție de vârstă. În primele 6 luni de viață, herniile inghinale, infecțiile respiratorii anormal de frecvente, otita și organomegalia pot fi observate la pacienții cu MPS, în special la MPS I (sindromul Hurler) care are cel mai precoce debut sever. Mai mult, de la 6 la 12 luni, acești sugari dezvoltă foarte des un gibbus (cifoză toraco-lombară), murmur cardiac, hernie ombilicală, hipotonie ușoară și întârziere de creștere . Ele pot avea, de asemenea, aspectul facial tipic grosier (Fig. 1). MPS II, VI și VII pot prezenta un fenotip timpuriu similar. Pacienții cu MPS IV au un fenotip scheletic timpuriu cu apariția displaziei șoldului în primele luni de viață și piept de porumbel (pectus carinatum) în primele 12-18 luni. Sugarii MPS II prezintă o implicare similară, dar cu debut ulterior. În al doilea an de viață, sugarii cu sindrom MPS I Hurler dezvoltă întârziere cognitivă, în timp ce un pacient MPS II ar putea fi identificat doar din cauza creșterii excesive, a infecțiilor frecvente ale căilor respiratorii și a intervențiilor chirurgicale multiple ; trăsăturile faciale tipice apar abia mai târziu. Unii pacienți cu MPS II dezvoltă, de asemenea, hiperactivitate între 18 și 24 de luni. În al treilea an de viață, hiperactivitatea și întârzierea cognitivă sunt ușor de recunoscut în MPS II și MPS III.
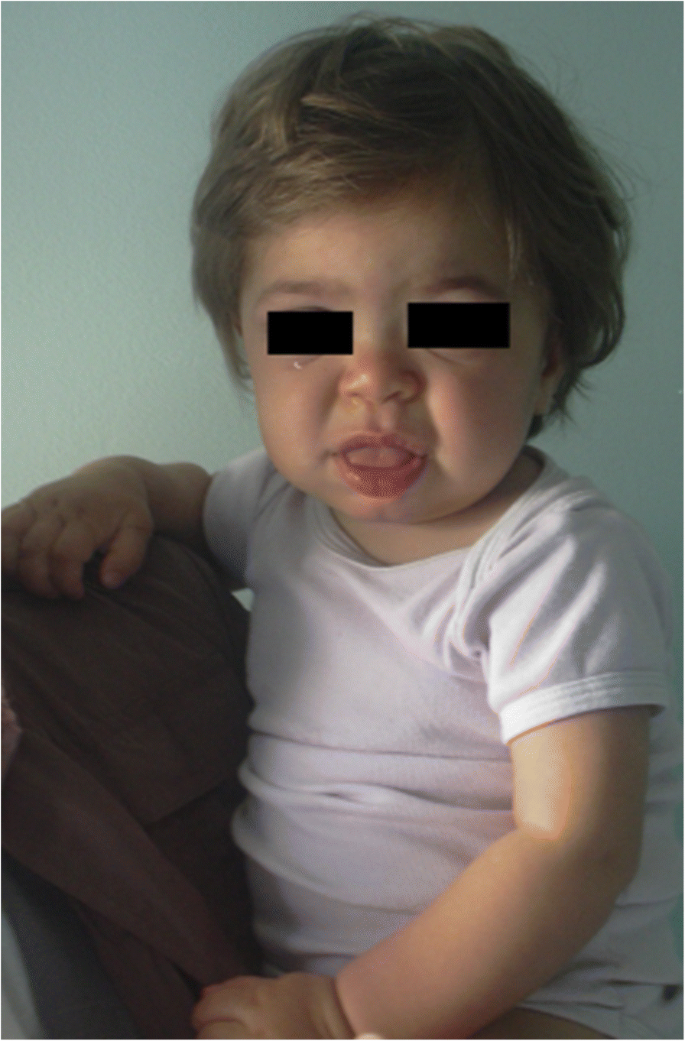
MPS IH la pacientul de 1 an. Facies grosiere: punte nazală plată, macroglossie, bossing frontal
pe scurt, manifestările scheletice sunt tipice și uneori precoce. Faciile grosiere și întârzierea cognitivă nu sunt semne proeminente timpurii.
care sunt diferitele prezentări ale diferitelor tipuri de deputați?
MPS cu implicare somatică și cognitivă
mucopolizaharidoză de tip I (MPS I; OMIM #252800) (Fig. 1, 2, 3, 4 și 5) este cauzată de o deficiență a hidrolazei lizozomale-l-iduronidazei, ceea ce duce la acumularea multisistemică a două gag: sulfat dermatan (DS) și sulfat heparan (HS) . Pe baza severității sale, MPS I este în mod tradițional împărțit în trei fenotipuri clinice; cu toate acestea, aceste forme reprezintă un continuum al severității bolii. Sindromul Hurler, cea mai severă formă, se prezintă de obicei în primul an de viață. Copiii afectați dezvoltă rapid tulburări cognitive semnificative și boli somatice în mai multe sisteme de organe, ducând la deces în primul deceniu în absența tratamentului. Formele atenuate ale MPS I, cunoscute sub numele de sindromul Hurler–Scheie și, respectiv, sindromul Scheie, se caracterizează prin apariția ulterioară a simptomelor, speranța de viață mai lungă și implicarea ușoară sau deloc a sistemului nervos central (SNC). Moștenirea este autosomală recesivă și, în cel puțin 50% din cazuri, predicția fenotipului este posibilă pe baza genotipului, în timp ce în restul de 50% sunt detectate mutații noi . Prevalența este de aproximativ 1 din 100.000 de nașteri vii .

MPS IH. un Gibbus la un pacient de 9 luni. B coloana vertebrală X-ray a coloanei vertebrale la un pacient de 3 ani. c T2-imagine de rezonanță magnetică ponderată a coloanei vertebrale care prezintă cifoză semnificativă și stenoză a canalului vertebral

MPS IH. Dovezi cu raze X ale disostozei multiplex. o îngroșare a coastelor la un pacient în vârstă de 3 ani și displazie de șold b la un alt pacient în vârstă de 18 luni

MPS IH: o mână cu gheare la un pacient de 18 luni; B radiografie de mână.

MPS IH. o îngroșare a osului cortical al craniului și conformația anormală “în formă de J” a sella turcica. Dilatarea spațiilor periventriculare în imaginile de rezonanță magnetică ponderată b și c T-2 ale unui pacient în vârstă de 17 luni
mucopolizaharidoza de tip II (MPS II; OMIM # 309900) (Fig. 6), cunoscut și sub numele de sindromul Hunter, este rezultatul deficienței enzimei iduronat-2-sulfatază (I2S) cu acumularea GAG în consecință a DS și HS . Prevalența este de aproximativ 1 din 140.000–156.000 de nașteri vii de sex masculin . În special, acesta este singurul MPS legat de X, iar purtătorii de sex feminin sunt de obicei asimptomatici; cu toate acestea, pot fi afectați în mod excepțional din cauza anomaliilor cromozomului X, homozigozității sau inactivării x înclinate . Expresia fenotipică se întinde, de asemenea, pe un spectru larg de severitate clinică. În cazurile severe, implicarea neurologică progresivă marcată și boala somatică coexistă, iar moartea apare de obicei în a doua decadă a vieții. Alți pacienți cu disfuncție neurologică minimă sau fără disfuncție neurologică pot prezenta o implicare somatică severă, în timp ce, la capătul opus al spectrului, există pacienți cu manifestări somatice minime și inteligență normală care supraviețuiesc până la vârsta adultă . Semnele și simptomele timpurii sunt împărtășite de formele severe ale MPS I și II.

MPS II. Un pacient în vârstă de 3 ani, cu trăsături faciale caracteristice ușoare
cazurile sporadice de hydrops fetalis sunt descrise în MPS I, deși, în general, acești copii par normali la naștere. Primele semne ale parlamentarilor apar de obicei în primele luni de viață, în timp ce cele din parlamentarii II apar de obicei puțin mai târziu . Kiely și colab. , într-un studiu retrospectiv la 55 de pacienți Hurler, a observat că simptomele respiratorii, dificultățile de hrănire, hernia inghinală și otita medie au apărut la o vârstă medie de 6 luni, iar cifoza, anomaliile cardiace, circumferința capului mărită, hipotonia, organomegalia, înnorarea corneei, restricția articulară și hernia ombilicală au apărut între 6 și 12 luni. Hernia ombilicală este una dintre cele mai vechi caracteristici care prezintă MPS I și II, iar herniile inghinale sunt raportate la aproximativ 60% dintre pacienți .
un alt indiciu inițial comun pentru MPS I și II poate fi infecțiile recurente ale căilor respiratorii superioare cu secreții crescute. Otita frecventă, rinita cronică recurentă și secreția nazală persistentă fără infecție evidentă nu sunt specifice, dar pot ajuta la întărirea suspiciunii diagnostice dacă sunt asociate cu semne mai specifice. Depozitarea GAG în oro-faringe duce la mărirea amigdalelor și a adenoidelor și contribuie la complicațiile căilor respiratorii superioare. O altă constatare frecventă este respirația zgomotoasă, în special noaptea, asociată în etapele ulterioare cu apneea obstructivă a somnului. Ca o consecință a infecțiilor frecvente ale urechii medii și a disostozei osicolelor urechii medii, pierderea auzului este frecventă, cu deficite conductive și senzorineurale . Traheobronchomalacia este frecvent observată și poate duce la obstrucție acută a căilor respiratorii sau colaps, aceasta fiind una dintre principalele cauze de deces în anii următori .
adenoidectomia, amigdalectomia și timpanostomia, împreună cu repararea herniei, sunt intervențiile chirurgicale mai frecvente și mai timpurii pentru pacienții MPS I și II, adesea efectuate înainte de a cunoaște diagnosticul (în mai mult de 50% din cazuri) .
diareea recurentă este destul de frecventă în stadiile incipiente ale MPS I și poate fi prezentă și la pacienții cu MPS II cu implicare neurologică, în timp ce este mai puțin frecventă la pacienții ușor afectați .
deformarea Gibbusului (cifoză dorso-lombară) (Fig. 2) devine evident clinic la vârsta medie de 8 ani.6 luni la 1 an în MPS I, reprezentând un marker destul de specific și precoce al MPS în general. Corpurile vertebrale apar aplatizate și cioc, ceea ce poate duce la complicații ulterioare, cum ar fi prinderea nervului spinal, leziuni acute ale coloanei vertebrale și instabilitate atlanto-occipitală; astfel, trebuie acordată o atenție deosebită prevenirii dislocării articulației atlanto-axiale în timpul anesteziei generale și al intervenției chirurgicale. După vârsta de 1 an, contracturile articulare și genu valgum reprezintă alte semne frecvente de afectare articulară și displazie scheletică progresivă, care sunt ușor detectabile la vârsta mai devreme de 2 ani la toți sugarii afectați de MPS. Toate aceste manifestări scheletice împreună, tipice MPS, sunt numite disostoză multiplex. Îngroșarea coastelor poate fi văzută pe radiografii (Fig. 3a), oasele lungi sunt scurte cu arbori largi, iar genunchii sunt predispuși la deformări valgus și varus. Disostoza falangiană și îngroșarea sinovială duc la o deformare caracteristică a mâinii ghearelor (Fig. 4), care este o altă caracteristică tipică care poate ridica suspiciunea bolii. Displazia de sold este frecvent motivul pentru care acesti copii pot fi vazuti pentru prima data de catre specialistii ortopedici. De obicei, pelvisul este slab format, capetele femurale sunt mici, iar coxa valga este comună, cu deformare progresivă și debilitantă a șoldului (Fig. 3b).
există o diferență notabilă între MPS I și MPS II; la MPS I, creșterea scheletului decelerează cu vârsta de 3 ani , în timp ce pacienții MPS II prezintă o creștere excesivă în primii 5-6 ani, încetinind ulterior creșterea lor .
coagularea trăsăturilor faciale (nasul larg cu nări evazate, crestele supraorbitale proeminente, obrajii mari rotunjiți, buzele groase, limba proeminentă mărită) cauzată de depozitarea gagurilor în țesuturile moi ale regiunii orofaciale și oasele faciale devine evidentă în primii 2 ani, la o vârstă medie de 1,1 ani pentru boala Hurler și între 18 luni și 4 ani în forma severă a bolii Hunter , deși modificările fenotipice subtile pot fi detectate mai devreme (încă din a doua jumătate a primului an) prin pediatri cu experiență specifică (fig. 1). Macrocefalia devine progresiv mai evidentă, iar scafocefalia este comună (Fig. 5), dar microcefalia este posibilă și la pacienții cu Hurler . Hirsutismul Facial și corporal sunt întotdeauna evidente până la vârsta de 24 de luni . Pielea poate fi îngroșată și inelastică. În special, unii pacienți cu MPS II dezvoltă o leziune distinctă a pielii, care este descrisă ca papule albe de Fildeș (asemănătoare pietricelelor) pe partea superioară a spatelui și pe părțile laterale ale brațelor superioare, patognomonice ale sindromului Hunter .
cardiomiopatia și îngroșarea progresivă și rigidizarea pliantelor valvei la pacienții cu MPS I pot duce la regurgitare mitrală și, mai puțin frecvent, aortică . Un murmur poate fi detectat în primele luni de viață și uneori poate fi raportat ca primul semn al bolii . Implicarea cardiacă este prezentă și la aproape toți pacienții cu MPS II, dar aceasta începe la vârsta de aproximativ 5 ani . Boala valvei poate duce la hipertrofie ventriculară dreaptă și stângă și insuficiență cardiacă.
protuberanța abdomenului cauzată de hepatosplenomegalia progresivă este ușor evidentă după vârsta de 6 luni, deși depozitarea gagurilor în ficat și splină nu duce la disfuncții ale organelor .
încețoșarea corneei apare la aproape toate persoanele cu MPS I la vârsta mai devreme de 15 luni , provocând potențial tulburări vizuale severe. Dimpotrivă, înnorarea corneei nu este o caracteristică tipică a sindromului Hunter , dar examinarea cu lampă cu fantă poate dezvălui leziuni corneene discrete care nu afectează vederea. Disfuncția retiniană poate fi detectată, în timp ce glaucomul nu este o constatare obișnuită.
dezvoltarea psihomotorie timpurie este în general detectată ca normală, dar în MPS i întârzierea dezvoltării este de obicei evidentă până la vârsta de 18 luni, cu un declin mental progresiv care duce la o dizabilitate intelectuală severă. Competențele lingvistice sunt foarte limitate la acești copii. O întârziere globală a etapelor de dezvoltare în MPS II devine evidentă începând cu 2 ani . Cu toate acestea, principala caracteristică neurologică a bolii severe Hunter este reprezentată de tulburări comportamentale marcate, cum ar fi hiperactivitatea, încăpățânarea și agresivitatea, care de obicei nu sunt observate la cei cu fenotip atenuat .
mucopolizaharidoza de tip VII (MPS VII; OMIM #253220), cunoscută și sub denumirea de sindrom Sly, este un MPS ultra-rar caracterizat prin activitatea deficitară a glucuronidazei de tip VIII, cu stocare lizozomală a sulfatului de condroitină (CS), DS și HS, ceea ce duce la disfuncții celulare și de organe. Primul pacient a fost descris de Sly și colab. în 1973 și un total de 145 de pacienți au fost raportați în literatura de specialitate până în prezent.
prezentarea clinică și progresia bolii MPS VII se întinde pe un spectru larg de severitate, de la manifestări precoce, severe, multisistemice și deces în primele luni, până la un fenotip mai blând cu debut ulterior, inteligență normală sau aproape normală și supraviețuire mai lungă .
caracteristicile fenotipului pacienților cu MPS VII seamănă cu cele ale MPS I și MPS II (statură scurtă, displazie scheletică, macrocefalie, hipertrofie gingivală, infecții recurente ale urechii, hepatosplenomegalie, hernii, afectare cardiacă, scăderea funcției pulmonare și insuficiență cognitivă). O proporție neașteptat de mare de pacienți descriși (41%) au antecedente de hidrops fetalis neonatal neonatal (NIHF) . În ciuda debutului prenatal al bolii la acești pacienți, 13 din 23 au supraviețuit copilăriei cu un curs ușor până la intermediar până la adolescența târzie. Astfel, prezența NIHF nu prezice, prin ea însăși, posibila severitate a cursului clinic dacă pacientul supraviețuiește copilăriei. Un alt semn precoce este macrocrania, în timp ce implicarea valvei cardiace și infecțiile repetate ale tractului respirator superior și inferior apar în primii 2 ani de viață. Întârzierea mentală moderată și pierderea progresivă a auzului cu tulburări de vorbire sunt, de asemenea, evidente în timp.
în rezumat, formele severe de MPS am un debut multisistem foarte timpuriu (în termen de 6 luni de viață); MPS II este similar, dar cu un debut ulterior, iar MPS VII are un debut prenatal cu nihf frecvent și implicare severă timpurie a tractului respirator superior și inferior.
MPS cu implicare neurologică și cognitivă
Mucopolizaharidoze de tip III (MPS III, cunoscut și sub numele de sindromul Sanfilippo; Fig. 7) are o prezentare neurologică predominantă. MPS III este o tulburare de stocare lizozomală cauzată de deficiența uneia dintre cele patru enzime implicate în catabolismul HS. Sunt cunoscute patru subtipuri diferite—MPS III tip A (OMIM #252900), tip B (OMIM #252920), tip C (OMIM #252930) și tip D (OMIM #252940)—fiecare datorită unei deficiențe enzimatice diferite: tip A, sulfamidază/gena sgsh; tip B, alfa-N-acetilglucosaminidază/gena NAGLU; tip C, alfa-glucozaminidă n-acetiltransferază/gena HGSNAT; Tipul d, gena n-ACETILGLUCOSAMINA-6-solfato sulfatasi/GNS). Toate cele patru tipuri (de la A la D) au moștenire autosomală recesivă. MPS III este cel mai frecvent dintre MPS cu o prevalență estimată între 0,3 și 4.1 cazuri pentru fiecare 100.000 de nou-născuți, în funcție de subtip și rasă .

MPS III. Același pacient la 2,5 și b 5 ani. Rețineți expresia diferită a feței, care arată progresia afectării neurologice
pacienții cu sindrom Sanfilippo prezintă tulburări cognitive progresive în al doilea și al treilea an de viață. Spre deosebire de majoritatea parlamentarilor, caracteristicile somatice sunt relativ mai puțin evidente, iar implicarea SNC este vizibilă. Întârzierea dezvoltării vorbirii este de obicei primul semn observat de părinți, dar principalele preocupări pot fi problemele comportamentale (hiperactivitate, comportament anxios și agresiv, tulburări de somn), urmate de declinul mental progresiv . Aceste simptome pot provoca diagnostic greșit ca tulburări de comportament, tulburare de hiperactivitate cu deficit de atenție (ADHD) și tulburări din spectrul autismului . Epilepsia poate fi, de asemenea, prezentă.
tulburările de somn, a căror incidență este raportată până la 80-90% , sunt o caracteristică comună. Acestea constau în dificultăți de adormire și trezire nocturnă frecventă, cu inversarea completă a ritmului zi–noapte la unii pacienți.
toate aceste manifestări sunt foarte greu de gestionat și au un impact psihologic enorm asupra calității vieții pentru întreaga familie.
în general, deși fenotipul poate fi foarte similar în cele patru subtipuri, evoluția clinică în Sanfilippo B și C pare să fie caracterizată de un fenotip mai puțin sever .
simptomele non-neurologice sunt de obicei mai puțin pronunțate în MPS III decât în celelalte MPS. Infecțiile recurente ale urechii, nasului și gâtului sunt observate la o vârstă mai mică. Otita recurentă, defectele osiculei urechii medii și anomaliile urechii interne pot provoca surditate. Un exces de secreții groase și modificări anatomice poate produce obstrucția căilor respiratorii. În primii ani de viață, diareea este frecvent raportată, în timp ce constipația este mai frecventă la pacienții vârstnici.
examenul fizic poate prezenta trăsături faciale grosiere, deși acestea sunt mai puțin pronunțate. Pacienții au macrocefalie, sprâncene largi cu flăcări mediale și sinofrii, părul este de obicei uscat și grosier, iar hipertricoza este frecventă. La pacienții mai tineri, se observă frecvent hepatomegalie ușoară și hernii ombilicale și inghinale. Contracturile Rare și adesea ușoare se găsesc în cea mai mare parte în coate. Creșterea este afectată la aproximativ jumătate dintre pacienții cu vârsta peste 12 ani .
în rezumat, boala MPS III sau Sanfilippo are o implicare neurologică proeminentă cu semne somatice foarte ușoare. Copiii cu vârsta cuprinsă între 2 și 3 ani care dezvoltă hiperactivitate, ADHD și comportament agresiv pot fi suspectați de a avea MPS III.
MPS cu implicare somatică predominantă
MPS IV și MPS VI prezintă doar implicare somatică. Acestea sunt condiții progresive care economisesc capacitatea intelectuală și afectează în principal scheletul (MPS IVA) sau toate organele/sistemele corpului (MPS VI). Moștenirea este autosomală recesivă. Rata la care simptomele se agravează variază în rândul persoanelor afectate.
mucopolizaharidoza IV (MPS IV, cunoscută și sub denumirea de sindrom Morquio; Fig. 8) se prezintă ca două tulburări distincte genetic, fiecare cu o deficiență enzimatică diferită: MPS IVA (sindromul Morquio A; OMIM #253000) datorită deficienței n-acetilgalactozamin-6-sulfatazei (gena GALNS), rezultând acumularea de keratan-sulfat (KS) și CS ; și MPS IVB (sindromul Morquio B; OMIM #253010) datorită deficienței activității beta-galactozidazei care duce la excreția de KS și oligozaharidă în urină ca la pacienții cu GM1. MPS IVB este într-adevăr alelic la diferitele forme de GM1-gangliosidoză, dar nu are deteriorarea psihomotorie observată în gangliosidoza GM1 .

MPS IVA. un pacient în vârstă de 4 ani cu raze X B antero-posterioare și C laterale care prezintă coaste “în formă de paletă” și vertebre aplatizate și rotunjite cu aspect tipic de ” ciocănire anterioară
pacienții cu MPS IVA sever, fără activitate enzimatică detectabilă, își dezvăluie primele simptome, de obicei în primul an de viață, deși sunt rareori diagnosticați înainte de vârsta de 4-5 ani. Informațiile privind istoricul natural al pacienților cu Morquio A provin în principal din două colecții largi de date . Rata de creștere redusă începe la vârsta de aproximativ 18 luni, iar creșterea se va opri la vârsta de aproximativ 7 sau 8 ani. Spre deosebire de celelalte MPS, articulațiile Morquio A sunt de obicei laxe și foarte flexibile (hipermobile), dar unele articulații (de obicei șoldurile și umerii) pot avea o gamă restrânsă de mișcare. Pacienții cu o astfel de implicare scheletică pot primi un diagnostic sau pot fi supuși unei evaluări pentru displazia spondiloepifizală, pseudoachondroplasia, displazia epifizală multiplă sau boala bilaterală Legg-Calv-Perthes .
o caracteristică constantă este hipoplazia odontoidă; astfel, dislocarea articulației atlanto-occipitale poate apărea provocând compresie și deteriorarea bulbarului și măduvei spinării, ducând la paralizie sau chiar moarte dacă stabilizarea cervicală nu este efectuată devreme.
pacienții cu MPS IVB au o manifestare clinică foarte asemănătoare, dar cu o statură scurtă mai puțin proeminentă. Au trăsături faciale ușor grosiere, pierderea auzului, opacități corneene, boli cardiace valvulare și infecții frecvente ale tractului respirator superior. De asemenea, pot prezenta hernie inghinală și hepatomegalie ușoară. Caracteristicile displazice scheletice includ platyspondyly, hipoplazie odontoidă cu posibilă subluxație cervicală, cifo-scolioză, coxa valga și aripi iliace restrânse .
mucopolizaharidoza VI (MPS VI, cunoscută și sub numele de sindromul Maroteaux-Lamy; OMIM #253200; Fig. 9) se datorează mutațiilor patogene ale genei ARILSULFATAZEI B (ARSB) situate pe cromozomul 5. În consecință, activitatea redusă sau absentă a enzimei ARILSULFATAZĂ B (ASB) (N-acetilgalactozamină 4-sulfatază) afectează degradarea DS determinând acumularea sa celulară .

MPS VI. un băiat de 2 ani cu o față grosieră evidentă și disostoză scheletică
pacienții fără activitate enzimatică detectabilă au forma severă de MPS VI și prezintă simptome în copilăria timpurie. Fenotipul lor somatic este similar cu sindromul Hurler. În cele mai multe cazuri, până la vârsta de 2 sau 3 ani, leziunile scheletice severe și progresive, numite disostoză multiplex, devin evidente. Această displazie scheletică include displazia șoldului cu cap femural displazic, corpuri vertebrale anormale, clavicule neregulate, ulna și raza distală hipoplazică și oase metacarpiene displazice și scurte; oasele carpiene și tarsale sunt hipoplazice și au un profil neregulat. Viteza de creștere încetinește adesea după primul an de viață, cu o înălțime finală în general mai mică de 120 cm. Ca și în cazul altor parlamentari, un semn timpuriu este trăsăturile faciale grosiere. În plus, pacienții au un abdomen tipic proeminent, hepatomegalie, hernie ombilicală și/sau inghinală și implicarea inimii în copilăria timpurie. Deși nu este prezentă nicio dizabilitate intelectuală primară, hidrocefalia poate fi observată la unii pacienți cu hipertensiune intracraniană și papiledem. Pectus carinatum, articulațiile rigide și contractate, scolioza și cifoza și compresia măduvei spinării cervicale se agravează cu timpul .
în rezumat, MPS IV și VI nu prezintă implicarea SNC. MPS IV este un MPS unic în manifestarea laxității comune, în timp ce debutul sever al MPS VI este similar cu cel observat la MPS I.