Autosomal-rezessive angeborene Ichthyose / Actas Dermo-Sifiliográficas
Einleitung
Die neueste Konsensus-Klassifikation der Ichthyose unterscheidet zwischen 2 Hauptformen: den nicht-syndromalen Formen, die nur mit Hauterscheinungen auftreten, und den syndromischen Formen, die auch mit Manifestationen in anderen Organen auftreten (Tabelle 1).1 Unter den nicht-syndromen Formen werden 4 Gruppen identifiziert: häufige Ichthyosen, autosomal-rezessive kongenitale Ichthyosen (ARCIs), keratinopathische Ichthyosen und andere weniger häufige Ichthyosen.Traditionell wurde die Gruppe der ARCIs in 2 Erkrankungen unterteilt, lamellare Ichthyose (LI) und kongenitale ichthyosiforme Erythrodermie (CIE). In der neuen Klassifikation wurde Harlekin-Ichthyose (HI) zu dieser Gruppe hinzugefügt1 weil inaktivierende Mutationen im ABCA12-Gen als verantwortlich für diese Störung identifiziert wurden,2,3 während Nonsense-Mutationen im selben Gen den Phänotyp LI4 oder CIE5,6 hervorrufen können. Andere weniger häufige Varianten, die in der Gruppe der ARCIs enthalten sind, sind selbstheilendes Kollodium-Baby (SHCB), akrales SHCB und Badeanzug-Ichthyose.7-9
Konsensus-Klassifikation basierend auf den klinischen Merkmalen der Ichthyosis1.
| Nicht-syndromale Formen | Syndromale Formen |
| Gemeinsame IchthyosesIchthyosis vulgarisRezessive x-chromosomale ichthyosis (nonsyndromic)ajimajor Formsharlekin ichthyosisLamellar ichthyosisCongenital ichthyosiform erythrodermaMinor Formsselbstheilendes Kollodium babyAcral self-healing collodium Babybadeanzug ichthyosisKeratinopathic IchthyosesMajor formsEpidermolytic Ichthyosissuperficial epidermolytic ichthyosisminor formenannuläre epidermolytische Ichthyosiscurth-Macklin-Ichthyosisautosomal-rezessiv epidermolytische Ichthyosisepidermolytischer Nävusandere Formenloricrin keratodermaErythrokeratodermie Vararabilispeeling-Hautsyndromangeborene retikuläre ichthyosiforme erythrodermaKLICK-Syndrom | Syndromische X-chromosomale Ichthyosrezessive x-chromosomale Ichthyose (syndromisch)Ichthyosis follicularis, Alopezie und Photophobie (IFAP) Syndromconradi-Scammann-Happle-Syndrom (chondrodysplasia punctata Typ 2)syndromische autosomale Ichthyosehautstörungenetherton-Syndromchthyose-Hypyrichose-Syndromchthyose-Sklerosierendes Cholangitis-Syndromrichothystrophyneurologische Störungenssjögren-Larsson syndromeRefsum diseaseMEDNIK syndromeFatal disease courseGaucher disease, type 2Multiple sulfatase deficiencyCEDNIK syndromeARC syndromeOther associated signsKID syndromeChanarin-Dorfman syndromeIchthyosis prematurity syndrome |
Abbreviations: ARC, arthrogryposis–renal dysfunction–cholestasis; ARCI, autosomal recessive congenital ichthyosis; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; KID, keratitis ichthyosis deafness; KLICK, keratosis linearis with ichthyosis congenital and sclerosing keratoderma; MEDNIK, geistige Behinderung, Enteropathie, Taubheit, periphere Neuropathie, Ichthyose, Keratodermie.
Zur Epidemiologie von ARCIs liegen nur begrenzte Daten vor. In den Vereinigten Staaten wurde eine Prävalenz bei der Geburt von 1 pro 100 000 Einwohner für LI und von 1 pro 200 000 Einwohner für CIE geschätzt. Andere Studien haben eine kombinierte Prävalenz für LI und CIE von 1 pro 200 000 bis 300 000 Einwohner berichtet.10,11 In einigen Ländern wie Norwegen ist die geschätzte Prävalenz aufgrund von Gründermutationen größer (1 pro 91 000).12 Der Befund von 1 oder mehreren wiederkehrenden Mutationen in einer Population kann darauf zurückzuführen sein, dass die Mutation zu einem bestimmten Zeitpunkt in der Geschichte aufgetreten ist und dann von Generation zu Generation weitergegeben wurde (Gründermutation) oder weil die Region des Genoms, in der die Mutation gefunden wird, eine DNA-Sequenz aufweist, die für Mutationen anfällig ist (Mutations-Hotspot). In Spanien beträgt die geschätzte Prävalenz von ARCI 1 pro 138 000 in der Allgemeinbevölkerung und 1 pro 61 700 bei Kindern unter 10 Jahren.13 In bestimmten Regionen Spaniens könnte die Prävalenz sogar noch höher sein. An der galizischen Küste wurde beispielsweise eine Prävalenz von 1 pro 33 000 gemeldet, was auch auf einen Gründereffekt zurückzuführen ist.14
Lamellare Ichthyose und kongenitales ichthyosiformes Erythrodermaklinische Merkmale
Obwohl ursprünglich angenommen wurde, dass LI und CIE unterschiedliche Entitäten sind, gab es Berichte über Patienten mit mittleren klinischen Manifestationen, und beide Zustände können durch Mutationen im selben Gen verursacht werden.15,16 Darüber hinaus können Patienten mit derselben Mutation, auch innerhalb derselben Familie, unterschiedliche Phänotypen entwickeln.12,15
Die meisten Patienten werden in einer Kollodiummembran geboren, die in den ersten Lebenswochen progressiv verschwindet und durch den endgültigen Phänotyp ersetzt wird (Abb. 1A). Hypohidrose, schwere Hitzeunverträglichkeit und Nageldystrophie werden sowohl bei LI als auch bei CIE häufig beobachtet.17-19 Patienten mit LI haben in der Regel schwerere klinische Manifestationen als Patienten mit CIE. Sie haben große plattenförmige Schuppen, oft von dunkler Farbe, die die gesamte Körperoberfläche bedecken. Erythrodermie ist entweder nicht vorhanden oder minimal. Solche Patienten haben normalerweise Ektropium und manchmal Eklabium, Hypoplasie des Gelenk- und Nasenknorpels, Narbenbildung, insbesondere am Rand der Kopfhaut, und palmoplantare Keratodermie (Abb. 1B und C). CIE ist durch das Vorhandensein von Erythrodermie und feiner weißlicher Schuppung gekennzeichnet (Abb. 2). Einige Patienten haben ein ausgeprägtes Erythem und eine generalisierte Schuppung. Die Schuppen können groß und dunkel gefärbt sein, insbesondere an den Streckseiten der Beine. In weniger schweren Fällen ist das Erythem mild und die Schuppung ist in Ordnung.

Klinische Merkmale der lamellaren Ichthyose. A, bräunliche lamellare Abschuppung. B, Markierte plantare Hyperkeratose. C, Narben Alopezie der Kopfhaut.
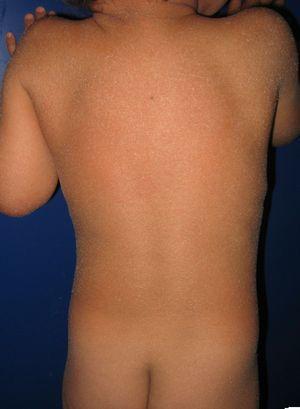
Patient mit kongenitaler ichthyosiformer Erythrodermie und Mutationen im ALOXE3-Gen. Ein leichtes Erythem und eine generalisierte weißliche Furchenschuppung sind zu sehen.
Histopathologie
Histopathologische Veränderungen liefern keine Diagnose. Bei LI wird eine massive orthokeratotische Hyperkeratose beobachtet, meist mit doppelter Ausdehnung wie bei CIE. Die Epidermis ist akanthotisch und nimmt gelegentlich ein Psoriasis-ähnliches Aussehen an. Die Zellproliferationsrate ist normal oder leicht erhöht.17-19 Patienten mit CIE haben eine weniger ausgeprägte Hyperkeratose mit fokaler oder ausgedehnter Parakeratose, einer normalen oder verdickten körnigen Schicht und einer ausgeprägteren Akanthose. Der epidermale Umsatz wird erhöht.17-19
Ultrastruktur
Obwohl eine enge Korrelation zwischen molekularen, klinischen und ultrastrukturellen Befunden bisher nicht gefunden wurde, kann die Elektronenmikroskopie dennoch nützlich sein, um andere Formen der Ichthyose auszuschließen und in einigen Fällen genetische Analysen zu leiten. Es wurden vier Arten von angeborener Ichthyose beschrieben (Tabelle 2).
Ultrastrukturelle Klassifikation angeborener Ichthyosen.
| Typ | Hauptmerkmal | Andere Merkmale | Mutationen | Klinische Manifestationen |
| 1 | Abwesenheit von ultrastrukturellen Markern der Ichthyose-Typen 2, 3 und 4 | Lipidtröpfchen oder Ringe im Stratum corneum (am häufigsten)Kleines Keratohyalingranulatvesikuläres oder lobuläres Membranbeschichtungsgranulat | TGM1 (33.3%)ALOX12B (2 Fälle) | CIE |
| 2 | Cholesterinspalten im Stratum corneum | Fehlen oder Ausdünnen verhornter Hüllenkleine Keratohyalingranulatlipidtröpfchen | TGM1 (89-100%) | LI |
| 3 | Laminierte Membranstrukturen im Stratum granulosum und/oder Stratum corneum. | Abnorme Membranbeschichtungsgranulatellipidtropfenfoki prominenter juxtanukleärer Vakuolen in der Granulatschicht | NIPAL4 (93%) | CIE (am häufigsten)LI |
| 4 | Trilamellare Membranpakete, die einige Zellen im Stratum granulosum und / oder Stratum corneum füllen | Abnormale Membranbeschichtungsgranulate | FTAP4 | Ichthyose-Frühgeburtssyndrom (100%) |
Abkürzungen: CIE, angeborene ichthyosiforme Erythrodermie; LI, lamellare Ichthyose.
Angeborene Ichthyose Typ 1
Angeborene Ichthyose Typ 1 ist durch das Fehlen ultrastruktureller Marker für die Ichthyose-Typen 2, 3 und 4 gekennzeichnet. Daher wird die Diagnose normalerweise nur gestellt, wenn die anderen Typen ausgeschlossen wurden. Der häufigste Befund ist das Vorhandensein von Lipidtröpfchen oder -ringen im Stratum corneum (Abb. 3A).20 Diese Lipidtröpfchen sind kein konstantes Merkmal oder spezifisch für diesen speziellen Typ, da sie nicht in allen Fällen vorhanden sind,20 und sie können bei anderen Arten von Ichthyose vorhanden sein.21,22 Klinisch weisen die meisten Patienten Manifestationen von CIE auf.12,20 Ein Drittel der Patienten hat Mutationen im TGM1-Gen.16 Dieser ultrastrukturelle Typ wurde auch in Verbindung mit Mutationen im ALOX12B-Gen identifiziert.23,24

Elektronenmikroskopische Aufnahmen. A, Angeborene Ichthyose Typ 1, zeigt Lipidtröpfchen im Stratum corneum und Fehlen von ultrastrukturellen Markern der anderen Arten von Ichthyose. B, Angeborene Ichthyose Typ 2, gekennzeichnet durch das Vorhandensein von Cholesterinspalten (Pfeil) in Korneozyten.
Angeborene Ichthyose Typ 2
Angeborene Ichthyose Typ 2 ist durch Cholesterinspalten im Stratum corneum gekennzeichnet (Abb. 3B).21 Solche Spalten sind ein ständiger Befund bei dieser Art von Ichthyose und können in verschiedenen Biopsien bei demselben Patienten nachgewiesen werden; Die Behandlung mit oralen Retinoiden hat keinen Einfluss auf diese Spalten.12,25 Elektronendichte Aggregate wurden auch auf Korneozyten bei einigen Patienten mit defizienter TGase 1-Aktivität beobachtet.26-28 Klinisch weisen die meisten Patienten schwere Manifestationen von CIE auf.12 Dieser ultrastrukturelle Typ ist stark mit Mutationen im TGM1-Gen assoziiert.12,16
Angeborene Ichthyose Typ 3
Angeborene Ichthyose Typ 3 ist durch lamellare Membranstrukturen im Stratum granulosum und / oder Stratum corneum gekennzeichnet. Diese Strukturen sind in Streifen um einen leeren Raum in der Nähe des Kerns angeordnet.22,29-31 Die klinischen Manifestationen bei diesem Typ unterscheiden sich von den anderen; Der Beginn der Ichthyose ist variabel, Desquamation und Erythem können fleckig oder generalisiert sein, und insbesondere die Biegungen sind betroffen. Mutationen im NIPAL4-Gen sind für 93% der Ichthyosen Typ 3 verantwortlich.32
Angeborene Ichthyose Typ 4
Charakteristischerweise sind bei angeborener Ichthyose Typ 4 einige Zellen im Stratum granulosum und Stratum corneum mit trilamellaren Membranpaketen gefüllt.33 Diese Befunde sind pathognomisch für das Ichthyose-Frühgeburtssyndrom, eine Erkrankung, die derzeit als syndromale Form der Ichthyose angesehen wird.34,35
Molekulare Studien
In genetischer Hinsicht sind die ARCIs sehr heterogen. Das TGM1-Gen ist mit den meisten Fällen assoziiert, aber Mutationen in 5 anderen Genen (ALOX12B, ALOXE3, NIPAL4, CYP4F22 und ABCA12) wurden berichtet. Fischer et al.36 untersuchten 520 Familien mit ARCI und identifizierten in 78% der Fälle Mutationen in mindestens 1 dieser Gene (TGM1 in 32%, NIPAL4 in 16%, ALOX12B in 12%, CYP4F22 in 8%, ALOXE3 in 5% und ABCA12 in 5%). In einer anderen Studie mit 250 Patienten mit ARCI unterschiedlicher Herkunft hatten 38% TGM1-Mutationen, 6, 8% ALOXE3-Mutationen und 6, 8% ALOX12B-Mutationen.37 In Galizien identifizierten wir Mutationen in den Genen TGM1, ALOX12B, ALOXE3, NIPAL4 und CYP4F22 in 75% der untersuchten Familien, aber die Verteilung der Mutationen war unterschiedlich.14 Das TGM1-Gen wurde in 68 mutiert.7% der Fälle, während das ALOXE3-Gen bei nur 1 Patienten mutiert war. Wir haben keine Mutationen in einem der anderen 3 untersuchten Gene nachgewiesen.
TGM1
Das TGM1-Gen befindet sich auf Chromosom 14q11.2 und hat 15 Exons (GenBank NM-000359.2). Es kodiert für das Enzym TGase 1, das eines der 3 TGase-Enzyme in der Epidermis ist.38 Dieses Enzym ist an der Bildung der verhornten Hülle beteiligt, indem es die kalziumabhängige Vernetzung mehrerer Proteine wie Involucrin, Loricrin und prolinreiche Proteine katalysiert.39,40 Es katalysiert auch die Bindung von ??-Hydroxyceramide in der äußeren Schicht der verhornten Hülle mit Proteinen in der inneren Schicht.41,42 Bei Patienten mit TGM1-Mutationen fehlt die verhornte Hülle und die TGase 1-Aktivität ist reduziert oder nicht vorhanden.43-47
Seit 1995, als dieses Gen als verantwortlich für einige Fälle von ARCI identifiziert wurde, wurden 48-50 mehr als 110 Mutationen bei Patienten unterschiedlicher Herkunft berichtet. Mutationen in TGM1 sind die häufigste Ursache für ARCI.36,37 Diese Mutation wurde in 55% der Fälle in den Vereinigten Staaten und in 84% der Fälle in Norwegen gefunden.12,51 Die häufigste Mutation ist c.877-2A>G, das in 34% der bisher gemeldeten mutierten Allele gefunden wurde.52 Die hohe Häufigkeit dieser Mutation in Ländern wie den Vereinigten Staaten und Norwegen ist auf einen Founder-Effekt zurückzuführen.12,53 Die zweithäufigste Mutation ist p.Arg142His. Diese und ähnliche Mutationen wurden in Ländern wie Ägypten, Deutschland, Finnland und den Vereinigten Staaten berichtet,15,49-51,54-56, und es scheint, dass dies Hotspot-Mutationen sind.57 Die p.Arg307Trp-Mutation ist in der japanischen Bevölkerung häufig.5 In Galizien, die p.1760x, c.1223_1227delACACA und c.984+1G>A-Mutationen in TGM1 wurden in 81,82% der Familien mit Mutationen in diesem Gen identifiziert, was auf einen Founder-Effekt hindeutet.14 Bestätigung dieser Hypothese wurde durch Haplotypstudie erhalten (Arbeit noch unveröffentlicht).
TGM1-Mutationen sind für die meisten Fälle von LI15 verantwortlich,27,44,46,56,58-63 und für einen kleinen Prozentsatz der Fälle von CIE.43,47,64,65 Solche Mutationen können auch zu anderen Formen von ARCI wie SHCB, akralem SHCB und Badeanzug-Ichthyose führen.
Viele Studien haben versucht, Genotyp-Phänotyp-Assoziationen zwischen Mutationen in TGM1 und ultrastrukturellen oder klinischen Befunden nachzuweisen, aber bisher wurde keine signifikante Korrelation beobachtet.15,16,53 Im Allgemeinen sind Patienten mit Mutationen im TGM1-Gen stärker betroffen als Patienten ohne solche Mutationen. In einer Studie mit 83 Patienten mit ARCI in Schweden und Estland war das Vorhandensein von Ektropium- und Kollodiumbabys mit TGM1-Mutationen assoziiert, während bei Patienten ohne Mutationen in diesem Gen eine höhere Erythemrate beobachtet wurde.66 Eine andere Studie zeigte, dass die Art der Skalierung der Hauptunterschied zwischen Trägern und Nichtträgern von TGM1-Mutationen ist, als festgestellt wurde, dass alle Patienten mit Mutationen in diesem Gen eine lamellare Skalierung aufwiesen, während 80% derjenigen ohne TGM1-Mutationen eine feine Skalierung aufwiesen.14 Darüber hinaus hat sich gezeigt, dass trunkierende Mutationen häufiger mit Hypohidrose und Schwitzstörungen assoziiert sind als Missense-Mutationen.51 In der nordamerikanischen Bevölkerung sagt ein Modell, das auf dem Vorhandensein bestimmter klinischer Merkmale basiert, voraus, dass Patienten, die als Kollodium-Babys geboren werden und Augenerkrankungen und / oder Alopezie haben, 4-mal häufiger TGM1-Mutationen aufweisen.51
ALOXE3 und ALOX12B
Die Gene ALOXE3 und ALOX12B befinden sich auf Chromosom 17p13.1.67 Sie haben eine ähnliche Struktur mit 15 Exons, die für die epidermalen LOXs eLOX-3 und 12R-LOX kodieren.68,69 Die Tatsache, dass sie überwiegend in den suprabasalen Schichten der Epidermis exprimiert werden, unterstützt ihre Rolle in fortgeschrittenen Phasen der epidermalen Differenzierung mit Beteiligung an der Verarbeitung von Lamellenkörpern.24,70 Diese Enzyme wirken auf benachbarte Schritte im Hepoxilinweg (Abb. 4). 12R-LOX wandelt Arachidonsäure in 12R-Hydroxyeicosatetraensäure um, während eLOX-3 dieses Produkt in ein Epoxyalkoholisomer69,71 der Hepoxilin A3-Familie umwandelt.72 Das Hepoxilinprodukt ist instabil und wird in Zellen zu einem spezifischen Trihydroxyderivat (Trioxilin) hydrolysiert. Obwohl die genaue Rolle der Produkte des Hepoxilin-Signalwegs nicht bekannt ist, wurde spekuliert, dass sie an der Bildung interzellulärer Lipide des Stratum corneum beteiligt sein oder als Signale zur Induktion der Keratinozytendifferenzierung wirken können.
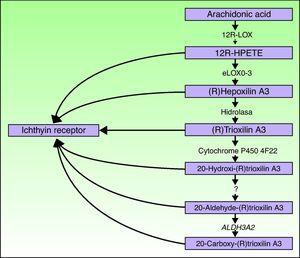
Schematische Darstellung des Hepoxilin-Signalwegs, die die Beteiligung der Gene ALOXE3, ALOX12B, NIPAL4 und CYP4F22 zeigt. Mutationen in diesen Genen sind für einige Arten von ARCI verantwortlich. HPETE zeigt Hydroperoxyeicosatetraensäure an.
Die Gene ALOX12B und ALOXE3 wurden erstmals 2002 identifiziert.73,74 Seitdem wurden mehr als 30 Mutationen im ALOX12B-gen23,24,37,75-77 und ungefähr 10 im ALOXE3-gen37,74,75 berichtet. Diese Mutationen sind für 14% bis 17% von ARCIs36,37 und 72 verantwortlich.2% von SHCBs.23,78,79 Die ursächliche Beziehung zwischen diesen Mutationen und dem Phänotyp wurde durch den Nachweis bestätigt, dass die katalytische Aktivität des epidermalen LOX bei Patienten mit diesen Mutationen vollständig aufgehoben wurde75,80, und durch die Verwendung von Tiermodellen, die den beim Menschen beobachteten ichthyosiformen Phänotyp reproduzierten.81-83 Beide Gene sind für einen ähnlichen Prozentsatz der ARCI-Fälle verantwortlich. Der Bereich der verschiedenen Mutationen im ALOXE3-Gen ist jedoch begrenzt, da 2 Mutationen, p.Arg234X und p.Pro630Leu, vorherrschen, die Hotspots zu entsprechen scheinen.37,74,75
Die Patienten mit Mutationen in den Genen ALOXE3 und ALOX12B zeigen in der Regel einen CIE-Phänotyp.74,75,77 Der Schweregrad der Schuppung ist mild oder mäßig, und die Schuppen haben eine weißliche oder hellbraune Farbe. Erythem kann ebenfalls vorhanden sein. Bis zu 76% der Patienten werden als Kollodium-Babys geboren und 88% haben Schwitzstörungen.37 Patienten mit Mutationen im ALOX12B-Gen zeigen im Vergleich zu Trägern von Mutationen im ALOXE3-Gen eine eingeschränktere, weißliche Abschuppung. In diesen Fällen sind die Schuppen bräunlich und anhaftend. Das Vorhandensein von Erythem, palmoplantärer Hyperkeratose und Akzentuierung der palmoplantaren Falten sind ebenfalls mit ALOX12B-Mutationen assoziiert.37
Ichthyin /NIPAL4
Das NIPAL4-Gen, auch als Ichthyin-Gen bekannt, befindet sich auf Chromosom 5q33. Es hat 6 Exons, die ein Protein mit mehreren Transmembrandomänen unbekannter Funktion kodieren.84 Es wurde die Hypothese aufgestellt, dass das Proteinprodukt am selben Stoffwechselweg wie LOX beteiligt ist und als Rezeptor für die Trioxiline A3 und B3 oder für andere Metaboliten des Hepoxilin-Stoffwechselwegs fungieren kann.84 Es wäre somit an der Bildung von Lamellenkörpern oder an deren Transport in den extrazellulären Raum beteiligt.32 Zur Unterstützung dieser sind 2 Beobachtungen. Erstens sind Mutationen in diesem Gen in 93% der Fälle mit einem ultrastrukturellen Muster der kongenitalen Ichthyose Typ 3 assoziiert, das durch Abnormalitäten in den Lamellenkörpern und das Vorhandensein länglicher perinukleärer Membranen im Stratum granulosum gekennzeichnet ist.32 Zweitens wird NIPAL4 im Wesentlichen im Stratum granulosum der Epidermis exprimiert, wo die Lamellenkörper vorhanden sind.85
Seit der Entdeckung des NIPAL4-Gens im Jahr 2004,84 wurden nur 9 Mutationen bei Patienten aus Mittelmeerländern (Algerien, Türkei und Syrien), 84 skandinavischen Ländern, 32 Pakistan, 85 den Färöern, 32 und Südamerika gemeldet.84
Das klinische Spektrum von Patienten mit Mutationen in diesem Gen ist breit, sogar unter Mitgliedern derselben Familie. Zwischen 3, 7% 32 und 60% 84 werden als Kollodiumbabys geboren. Wenn die Kollodiummembran verschwindet, entwickeln die meisten Patienten die Manifestationen von CIE mit feinen weißlichen Schuppen auf einer erythematösen Basis auf Gesicht und Rumpf und größeren, bräunlichen Schuppen an Hals, Gesäß und Beinen.84 Ausgeprägte Xerose, generalisierte bräunliche retikuläre hyperkeratotische Plaques, die in den Hautfalten akzentuiert erscheinen, und Gesichtsdyschromie können vorhanden sein.32,85 Darüber hinaus ist palmoplantare Keratodermie ein häufiger Befund zusammen mit gelegentlichen Fingerkontrakturen und gebogenen Fingernägeln. Einige Studien haben Ergebnisse berichtet, die für LI typischer sind.32,85 Bei einigen Patienten wurde über Anzeichen und Symptome einer atopischen Dermatitis berichtet, obwohl in keinem dieser Fälle Mutationen im FLG-Gen nachgewiesen wurden.85
CYP4F22
Das FLJ39501- oder CYP4F22-Gen befindet sich auf Chromosom 19p13.12.86 Es hat 12 Exons87 und codiert ein P450-Cytochrom, Familie 4, Unterfamilie F, Polypeptid 2, Homolog der Leukotrien-B4-ω-Hydroxylase (CYP4F2). Die durch das Produkt von FLJ39501 in der Haut katalysierte Reaktion und die Substrate dieser Reaktion können analog zu ihren bekannten Homologen CYP4F2 und CYP4F3 abgeleitet werden.88 Es wurde die Hypothese aufgestellt, dass CYP4F2 und CYP4F3 am Hepoxilinweg beteiligt sind, indem sie die Umwandlung von Trioxilin A3 in 20-Hydroxy-(R) trioxilin A387 katalysieren und dass das Endprodukt dieses Weges, 20-carboxytrioxilin A3, eine wichtige biologische Regulation bewirken kann Wirkung in der Haut.89
Bisher wurden nur 8 Mutationen dieses Gens in 12 blutsverwandten Familien aus Mittelmeerlanden87 und in 1 Familie israelischer Herkunft berichtet.62
In den von Lefèvre et al.,87 Die meisten Patienten hatten einen CIE-Phänotyp bei der Geburt und dieser entwickelte sich anschließend zu LI. Die Patienten wurden normalerweise mit ausgeprägter Erythrodermie geboren, jedoch ohne Kollodiummembran. Mit zunehmendem Alter entwickelten sie eine generalisierte weißlich-graue Schuppung, die im periumbilikalen Bereich, am Gesäß und am unteren Körperteil stärker ausgeprägt war. Hyperlinearität der Handflächen und Fußsohlen und Abschuppung auf der Kopfhaut, zu Zeiten des pityriasiformen Typs, waren häufig.87 In einer anderen Familie wurden die 3 betroffenen Mitglieder als Kollisionsbabys geboren und entwickelten intensive Erythrodermie, generalisierte Desquamation und palmoplantare Keratodermie.62
ABCA12
Im Jahr 2003 wurde berichtet, dass das ABCA12-Gen für einige Fälle von LI verantwortlich ist und auf Chromosom 2q34 abgebildet wurde.4 Anschließend wurde bestätigt, dass Mutationen in diesem Gen auch für HI verantwortlich waren.2,3ABCA12 kodiert für 53 Exons und gehört zu einer Familie von ABC-Transportern, die Adenosintriphosphat binden und gleichzeitig den Transport mehrerer Moleküle durch die Zellmembran erleichtern.90 Die Mitglieder der ABCA-Unterfamilie sind alle am Lipidtransport beteiligt.91 Eine mangelhafte ABCA12-Funktion verursacht Lipidtransportstörungen in Lamellenkörpern und führt so zu einer Abnahme der interzellulären Lipidspiegel im Stratum corneum.3ultrastrukturelle Studien haben gezeigt, dass sich ABCA12 in Lamellenkörpern befindet, die mit Glycosylceramiden assoziiert sind.91ABCA12-Mutationen wurden mit Störungen der Verteilung und des Transports von Glykosylceramiden und mit verminderten Spiegeln von Hydroxyceramiden, einer der Hauptkomponenten der Lipidbarriere in den Interzellularräumen, in Verbindung gebracht.3,6,92,93 Die massive Hyperkeratose, die bei diesen Patienten auftritt, könnte eine kompensatorische Reaktion auf eine mangelhafte Lipidbarriere sein.94 Es könnte auch an der fehlenden Abschuppung der Korneozyten liegen,93 die durch Defekte im Transport bestimmter Proteasen wie Callicrein 5 und Cathepsin D verursacht werden könnte, die aus Störungen in den Lamellenkörpern resultieren.95 Mausmodelle und In-vitro-Studien legen nahe, dass ABCA12-Mutationen auch einen Einfluss auf die epidermale Differenzierung haben.95-97
Bisher wurden mehr als 50 Mutationen im ABCA12-Gen bei Patienten mit ARCI aus Afrika, Europa, Pakistan und Japan berichtet. Die häufigsten Mutationen sind p.Val244SerfsTer28,2,98,99 identifiziert in pakistanischen und indischen Populationen und p.Asn1380Ser,4 identifiziert in afrikanischen Familien. In beiden Fällen kann es sich um genetische Mutationen handeln.
Das Ausmaß der ABCA12-Mutationen hängt mit dem Phänotyp zusammen, wobei Mutationen, die mit einem vollständigen Funktionsverlust verbunden sind, zum HI-Phänotyp führen.2,3,98-102 Im Gegensatz dazu sind in LI und CIE die meisten Mutationen missense und haben eine weniger schwere Wirkung auf die Proteinfunktion.4-6,103 Die dem LI-Phänotyp zugrunde liegenden Mutationen scheinen in der ersten Adenosintriphosphat-Bindungskassettenregion konzentriert zu sein.4 Klinisch haben Patienten mit CIE und Mutationen im ABCA12-Gen mittelgroße Skalen, die etwas größer sind als die, die normalerweise bei Patienten mit diesem Phänotyp beobachtet werden.
Harlekin-Ichthyose
HI oder Harlekin-Fötus ist eine schwere und meist tödliche Form der Ichthyose. Die Kinder sind in der Regel verfrüht mit ausgedehnten glänzenden hyperkeratotischen Plaques, die durch tiefe Risse getrennt sind, die das gesamte Integument bedecken und geometrische Muster bilden, die an Kleidung erinnern, die von Harlekinen getragen wird, wodurch der Zustand seinen Namen erhält. Die Enge der Haut führt zu einer deutlichen Umstülpung der Augenlider und Lippen, einer rudimentären Entwicklung des Gelenk- und Nasenknorpels und gelegentlich zu Mikrozephalie. Die Kinder haben selten Wimpern oder Augenbrauen, obwohl die Haare auf der Kopfhaut konserviert werden können. Die Hände und Füße sind geschwollen und ödematös und oft von einer handschuhartigen Schicht bedeckt. Sie können Fingerkontrakturen haben.
Für solche Patienten ist das Risiko, während der Neugeborenenperiode zu sterben, sehr hoch.104 Die Lungenventilation ist beeinträchtigt, der transepidermale Wasserverlust führt zu Dehydratation, hydroelektrischem Ungleichgewicht und thermischer Instabilität und das Infektionsrisiko ist erhöht. Gesichtsverspannungen und Eklabium behindern das Saugen und damit das Füttern mit der entsprechenden Verschlechterung der Dehydration. Neugeborene mit dieser Erkrankung lebten selten länger als ein paar Wochen. In den letzten Jahren haben sich die Überlebenschancen jedoch deutlich erhöht, was im Wesentlichen auf die Verabreichung von systemischen Retinoiden und Fortschritte in der Intensivpflege von Neugeborenen zurückzuführen ist.105 In einer kürzlich durchgeführten Studie überlebten 83% der mit oralen Retinoiden behandelten Patienten im Vergleich zu 24% der unbehandelten Patienten. Die meisten Todesfälle ereigneten sich in den ersten 3 Lebenstagen, bei vielen Überlebenden wurde jedoch erst danach mit der Behandlung begonnen.104 Dies würde darauf hindeuten, dass viele dieser frühen Todesfälle unabhängig von der Retinoidbehandlung aufgetreten wären.
Die Kinder, die die Neugeborenenperiode überleben, entwickeln im Allgemeinen eine schwere CIE.106 Die Art und der Ort von Mutationen im ABCA12-Gen und das Ausmaß des Transporterfunktionsverlusts können die Prognose bestimmen.3,92,107 Patienten, die ein gewisses Maß an Proteinaktivität bewahren, wenn auch minimal, können eine bessere Überlebenschance haben. Träger von homozygoten Mutationen haben eine höhere Sterblichkeitsrate.104
Das histologische Hauptmerkmal von HI ist das Vorhandensein eines extrem dicken und kompakten orthokeratotischen Stratum corneum. Die Haarfollikel und Schweißkanäle weisen prominente hyperkeratotische Pfropfen auf107,108 und haben abnormale oder fehlende Lamellenkörper, Lipideinschlüsse oder Reste von Organellen oder Kernen in den Korneozyten und Abwesenheit von interzellulären Lipiden in der ultrastrukturellen Studie.108,109 Die Haarfollikel zeigen eine ausgeprägte Konzentration an keratotischem Material, was ein diagnostisches Merkmal von HI ist, das für die Pränataldiagnostik verwendet wird.
Bis heute liegt die Nachweisrate von Mutationen im ABCA12-Gen bei Patienten mit HI nahe bei 100%, und dies scheint ein genetisch homogener Zustand zu sein.
Kollodiumbaby und selbstheilendes Kollodiumbaby
Kollodiumbabys werden normalerweise vorzeitig geboren und die perinatale Morbidität und Mortalität sind erhöht. Bei der Geburt ist das Neugeborene von einer glänzenden, transparenten Membran bedeckt, die an eine Zellophanverpackung erinnert (Abb. 5). Die Babys haben Ektropium, Eklabium und Hypoplasie des Nasen- und Gelenkknorpels. Das Saugen und die Lungenventilation können behindert werden110 und der transepidermale Wasserverlust und das Infektionsrisiko sind erhöht.110,111

Kollodiumbaby, das sich später zu einem lamellaren Ichthyose-Phänotyp entwickelte.
Collodium Baby ist die übliche Präsentation für HI und CIE. Autosomal dominantes LI, 112,113 Sjögren-Larsson-Syndrom, 110 Trichothyodystrophie, 114 juvenile Gaucher-Krankheit, 110 neutrale Lipidspeicherkrankheit, Conradi-Hünermann-Happle-Syndrom, Hays-Wells-Syndrom und ektodermale Dysplasie115 können gelegentlich auch als Kollodiumablagerungen auftreten. Die Membran verschwindet bei 10% bis 24% der Neugeborenen spontan und weicht einer völlig normalen Haut.110,116 In der Vergangenheit wurden diese Fälle als LI des Neugeborenen beschrieben,117 aber sie werden nicht als SHCB bezeichnet.118 Einige Autoren haben den Begriff selbstverbessernde Kollodium-Ichthyose vorgeschlagen, da viele dieser Patienten, wenn sie später im Kindesalter oder als Erwachsene erneut untersucht werden, einen unterschiedlichen Grad an Anhidrose und Hitzeunverträglichkeit sowie leichte Anzeichen von Ichthyose wie Xerose und feine Abschuppung aufweisen, insbesondere in den Achselhöhlen und im Hals.78
Weder optische Mikroskopie noch ultrastrukturelle Untersuchungen von Kollodiumverbindungen sind spezifisch. Es ist daher vorzuziehen, die Hautbiopsie zu verzögern, bis sich der endgültige Phänotyp entwickelt hat.
Mutationen in den Genen TGM1,7,119ALOXE3,78 und ALOX12B23,78,79 wurden bei Patienten mit SHCB identifiziert. ALOX12B-Mutationen sind die häufigsten. In einer Reihe von 15 skandinavischen Patienten mit SHCB hatten 67% Mutationen im ALOX12B-Gen, 25% im ALOXE3-Gen und 8,3% im TGM1-Gen.78 Mutationen wurden bei einigen Patienten nicht gefunden, so dass wahrscheinlich auch andere Gene beteiligt sind. Es wurde spekuliert, dass diese Mutationen die enzymatische Aktivität in der Gebärmutter verringern, jedoch nicht nach der Geburt.7 In der Gebärmutter, wo der hydrostatische Druck hoch ist, wandelt die Chelatbildung durch Wasser das mutierte Enzym in eine inaktive Konformation um. Nach der Geburt, wenn der Druck abnimmt, kehrt das Enzym in seine aktive Form zurück und seine Aktivität nimmt ausreichend zu, um einen normalen oder minimal betroffenen Phänotyp aufrechtzuerhalten.7
Akrales selbstheilendes Kollodium-Baby
Obwohl Kollodium-Baby den ganzen Körper betrifft, wurden Fälle berichtet, die auf die Akralregionen beschränkt sind. Im Jahr 1952 Finlay et al.120 berichteten von einem Fall von Kollodiummembran, der nur die Hände und Füße betraf und einem Selbstheilungskurs folgte. Kürzlich wurde ein neuer Fall von akralem SHCB in Verbindung mit Mutationen des TGM1-Gens berichtet.8 Es ist nicht bekannt, warum diese Läsionen auf akrale Regionen beschränkt sind, obwohl Faktoren, die mit einer ortsabhängigen Regulation der Enzymaktivität verbunden sind, in Betrieb sein können.8
Badeanzug-Ichthyose
Badeanzug-Ichthyose wurde erstmals 2005 als unabhängige ARCI-Variante gemeldet, obwohl zuvor Fälle von Ichthyose mit einer besonderen Verteilung gemeldet worden waren.121-123 Es wurde hauptsächlich bei Patienten südafrikanischer Herkunft nachgewiesen,9 obwohl es auch bei Personen aus Europa und den Mittelmeerländern berichtet wurde.124 Bei der Geburt haben die Patienten eine generalisierte Kollodiummembran, die sich dann ablöst, um die charakteristische Verteilung der Skalierung zu hinterlassen. Der Rumpf, die proximale Region der Arme einschließlich der Achselhöhlen, des Halses und der Kopfhaut sind im Allgemeinen betroffen, während der zentrale Teil des Gesichts, die Gliedmaßen und die Nebennierenregion normalerweise verschont bleiben.9 Die Schuppen sind groß, lamellar und dunkel gefärbt. Eine feinere Abschuppung kann in den Fossa poplitealis und antecubitalis auftreten.124,125 Die Handflächen und Fußsohlen weisen eine leichte diffuse Hyperkeratose auf, während der Hand- und Fußrücken keine Beteiligung aufweist.
Die histopathologische Untersuchung der betroffenen Haut zeigt eine ausgeprägte Hyperkeratose ohne Parakeratose, normale körnige Schichten, leichte oder mittelschwere Akanthose und ein leichtes lymphozytäres Infiltrat in der oberen Dermis.9 Elektronenmikroskopische Beobachtungen stimmen in den meisten Fällen mit kongenitaler Ichthyose Typ 2 überein. Unbeteiligte Haut zeigt keine auffälligen Befunde.124,125 Bei gesunder Haut ist die TGase 1-Aktivität leicht reduziert und normalerweise in perizellulären Bereichen lokalisiert. In der betroffenen Haut ist die enzymatische Aktivität restlich und abnormal im Zytoplasma lokalisiert.124
Bei allen bisher untersuchten Patienten mit Badeanzug-Ichthyose wurden Mutationen im TGM1-Gen nachgewiesen.119,124-126 Die häufigste Mutation ist p.Arg315Leu, die bei den meisten südafrikanischen Patienten identifiziert wurde und eine Gründungsmutation sein könnte. In: Oji et al.124 vorgeschlagen, dass die Hauttemperatur eine Rolle bei der Entwicklung dieser Manifestationen spielen könnte. Mithilfe der digitalen Thermografie zeigten die Autoren eine starke Korrelation zwischen Körpertemperatur und Abschuppung, wobei die heißesten Bereiche des Körpers am stärksten betroffen waren. Aufenvenne et al.127 zeigte eine Abnahme der optimalen Temperatur für die TGase 1-Aktivität bei Patienten mit Badeanzug-Ichthyose. Diese Abnahme wurde bei gesunden Kontrollen oder bei Patienten mit generalisierter LI nicht beobachtet. Die optimale Temperatur beträgt 37 ° C für das normale Enzym, aber 31 ° C für das mutierte Enzym.
Behandlung
Das Hauptziel der Behandlung bei Ichthyose ist die Beseitigung von Ablagerungen und die Verringerung der Xerose ohne übermäßige Reizung (Tabelle 3). Vor der Entscheidung über die Behandlung sollten Aspekte wie Alter und Geschlecht des Patienten, Art und Schwere der Erkrankung sowie Ausmaß und Ort der Läsionen berücksichtigt werden.128
Therapeutische Strategie bei autosomal-rezessiven angeborenen Ichthyosen.
| Therapeutische Strategie für die autosomal-rezessive angeborene Ichthyosen | |
| Baden und mechanische Beseitigung von Schuppen | Baden mit Natriumbicarbonat oder Weizenstärke, Maisstärke oder Reisstärke; mechanische Entfernung der Waage (1 oder 2 mal am Tag) |
| Topische Behandlung (sequentiell) | Harnstoffhaltige Feuchtigkeitsspenderkeratinolytika mit Propylenglykolkombinierte Keratinolytika (Propylenglykol, α-Hydroxysäuren oder Harnstoff)Keratinolytika in Kombination mit Salicylsäuretopische Retinoidebei Neugeborenen und Kleinkindern ein Vehikel ohne Wirkstoffe auftragen. Vermeiden Sie Harnstoff, Salicylsäure und Milchsäure aufgrund des Risikos einer systemischen Resorption |
| Orale Behandlung | Orale Retinoide (Acitretin oder Isotretinoin) |
| Andere Maßnahmen | Follow-up von Ektropium durch den Augenarztregelmäßige Reinigung des Außenohrs durch den Hals-Nasen-Ohrenspezialistenphysiotherapie zur Vorbeugung von Kontrakturen.Vermeidung von anstrengenden Tätigkeiten bei hohen Umgebungstemperaturenhydrotherapie |
Baden und mechanische Beseitigung von Schuppen
Patienten mit ARCI wird ein tägliches Baden empfohlen, um Schuppen und Spuren von Feuchtigkeitscreme mechanisch zu beseitigen. Dies ist einfacher, wenn der Patient 15 bis 30 Minuten in Wasser eingetaucht ist. Einige Autoren empfehlen, dem Bad Natriumbicarbonat zuzusetzen, um die Keratine zu denaturieren und das Wasser alkalisch zu machen und so die Beseitigung der Schuppen zu erleichtern.129 Andere Produkte, die hinzugefügt werden können, umfassen Weizenstärke, Maisstärke oder Reisstärke. Badeöle sind nicht geeignet, da sie zu einem Verschluss mit anschließendem Risiko einer bakteriellen Proliferation und einer Verschlechterung der Thermoregulation führen können.
Topische Behandlung
Feuchtigkeitscremes und topische Keratolytika sind normalerweise die erste therapeutische Option. Sie verbessern die Hautbarrierefunktion und erleichtern die Abschuppung. Leichte lokale Nebenwirkungen wie vorübergehender Juckreiz, Reizung oder Stechen können auftreten.
Natriumchlorid, Harnstoff, Vitamin E-Acetat, Glycerin und Vaseline können als Feuchtigkeitscremes und Gleitmittel verwendet werden. Bei Patienten mit dicker Schuppung und ausgeprägter Hyperkeratose können 1 oder mehr Keratolytika wie α-Hydroxysäuren (Milch- und Glykolsäure), 130 Salicylsäure, N-Acetylcystein,131-133 Harnstoff (> 5%),134 und Propylenglykol zugesetzt werden. Modulatoren der Keratinozytendifferenzierung werden ebenfalls verwendet. Dazu gehören topische Retinoide (Tretinoin, Adapalen, Tazaroten),135,136 Calcipotriol,137 und Dexpanthenol.Topische Retinoide verursachen oft Reizungen und kleine, sehr schmerzhafte Fissuren.137 Darüber hinaus besteht bei fruchtbaren Frauen das Risiko einer Resorption und Teratogenität, wenn sie zu intensiv angewendet werden.138 Um die Wirksamkeit von Keratolytika und Feuchtigkeitscremes zu verbessern, kann in bestimmten behandlungsresistenten Bereichen ein Okklusivverband angewendet werden.139 Ein additiver oder synergistischer Effekt kann auch durch die Kombination von 2 oder mehr Keratolytika oder Feuchtigkeitscremes erreicht werden.140-142 Die Behandlung sollte für jeden Einzelnen optimiert werden, da der Zustand und die Hautempfindlichkeit sehr unterschiedlich sind und die Reaktion auf jede Behandlung unterschiedlich ist. Der Optimierungsprozess kann unterstützt werden, indem eine Körperseite anders behandelt wird als die andere, um Vergleiche zu ermöglichen. Neugeborene und Kleinkinder sollten mit einem Vehikel ohne Wirkstoffe behandelt werden, da die Haut sehr fein und empfindlich ist und die meisten Keratolytika nicht vertragen werden. Darüber hinaus ist das Risiko einer perkutanen Resorption topischer Produkte wie Harnstoff, Salicylsäure und Milchsäure größer.143-145
Systemische Behandlung
Orale Retinoide haben keratolytische Wirkungen, die helfen, Schuppen zu beseitigen und übermäßige Hyperkeratose zu verhindern. Sowohl Isotretinoin als auch aromatische Retinoide (Acitretin und Etretinat) haben sich bei der Behandlung von ARCIs als wirksam erwiesen.128.146.147 Acitretin in einer Dosis von 0,5 bis 1 mg / kg / Tag ist das am häufigsten verwendete Medikament, insbesondere bei Patienten mit LI.148 Patienten mit CIE können eine vollständigere Reaktion und bei niedrigeren Dosen haben.
Die wichtigsten Nebenwirkungen sind mukokutane Störungen, Teratogenität, Erkrankungen des Bewegungsapparates sowie abnormales Lipidprofil und Transaminaseerhöhung.149-152 In Bezug auf die Teratogenität sollten die Arzneimittel im Fall von Etretinat und Acitretin während der Schwangerschaft vermieden werden, und die Patienten sollten vermeiden, 3 Jahre nach Absetzen der Behandlung schwanger zu werden.151 Isotretinoin hat eine kürzere Halbwertszeit und wird nach 1 Monat vollständig aus dem Organismus ausgeschieden und kann daher die bevorzugte Option bei Frauen sein, die schwanger werden möchten.128
Die Überwachung der Behandlung sollte eine Laboruntersuchung mit einem Leberfunktionstest und einem Lipidprofil vor Beginn der Behandlung, dann 1 Monat und alle 3 Monate nach Beginn der Behandlung umfassen. Bei fruchtbaren Frauen sollte in den 2 Wochen vor Beginn der Behandlung ein Schwangerschaftstest durchgeführt werden, und ab 4 Wochen vor der Behandlung bis 3 Jahre danach sollte eine wirksame Verhütungsmaßnahme angewendet werden (im Fall von Acitretin). Wenn eine längere Behandlung mit Retinoiden erforderlich ist, sollten Wachstum und Knochenentwicklung überwacht werden. Einige Autoren schlagen vor, vor der Behandlung eine Knochenstudie durchzuführen, gefolgt von einer jährlichen Untersuchung.151 Neuere Richtlinien empfehlen aufgrund der möglichen schädlichen Auswirkungen keine routinemäßige Radiographie.152 Stattdessen werden selektive Röntgenuntersuchungen bei Patienten mit atypischen Knochenschmerzen empfohlen.152
Eine Alternative zur systemischen Retinoidbehandlung ist die Verwendung von Arzneimitteln, die als Blocker des Retinsäurestoffwechsels bekannt sind und die endogenen Retinsäurespiegel erhöhen. Ein solches Medikament ist Liarozole, das von der Europäischen Arzneimittel-Agentur und der US-amerikanischen Food and Drug Administration den Orphan-Status für die Behandlung von LI, CIE und HI erhalten hat.153-155 Dieses Medikament hat sich in klinischen Studien als wirksamer als Acitretin erwiesen und ist auch besser verträglich und hat ein besseres pharmakokinetisches Profil.154
Sonstige medizinische Versorgung
Bei Patienten mit Ektropium kann die Anwendung künstlicher Tränen und Augenschmiermittel sowie die Befeuchtung der Haut des Gesichts und der Wangen insbesondere die palpebrale Retraktion verringern. Chirurgische Korrektur ist eine gültige Option in schweren Fällen, aber dies muss in der Regel ein paar Jahre später wiederholt werden. Hydrotherapie kann von Vorteil sein.156 Patienten sollten angewiesen werden, bei hohen Umgebungstemperaturen anstrengende körperliche Aktivitäten zu vermeiden, da eine Hypohidrose das Risiko eines Hitzschlags und von Krämpfen birgt. Orale Retinoide können die Thermoregulation verbessern.157 Physiotherapie ist wichtig, um Flexionskontrakturen zu verhindern, insbesondere bei HI. Eine regelmäßige Reinigung des äußeren Gehörgangs durch einen Hals-Nasen-Ohren-Spezialisten kann die Ansammlung von Schuppen und damit den Hörverlust verhindern.
Genetische Beratung und Pränataldiagnostik
Wenn bei einem Patienten Ichthyose diagnostiziert wird, sollte ihm eine geeignete genetische Beratung angeboten werden, in der die Art der Störung, der Übertragungsmodus und das Risiko zukünftiger Manifestationen in der Familie erläutert werden. Die Pränataldiagnostik kann anzeigen, ob der Fötus betroffen ist, und in diesem Fall kann eine psychologische Vorbereitung der Familie angeboten und Probleme während der Schwangerschaft und Geburt erwartet werden. Die Eltern können die Möglichkeit einer Abtreibung gegeben werden, wenn keine Behandlung zur Verfügung steht. Sollte in Zukunft eine Gentherapie für diese Erkrankungen verfügbar sein, würde die Pränataldiagnostik eine frühzeitige Anwendung dieser Therapie ermöglichen.
Seit mehr als 20 Jahren wurde die Pränataldiagnostik durchgeführt, indem eine Biopsieprobe der fetalen Haut entnommen und durch optische Mikroskopie, Elektronenmikroskopie oder Immunhistochemie untersucht wurde.158.159 Dieses invasive Verfahren konnte nur in den späten Schwangerschaftsphasen zwischen der 15. und 23. Schwangerschaftswoche durchgeführt werden und war mit einem Risiko von 1% bis 3% für den Verlust des Fötus verbunden.160,161 Die Identifizierung der molekularen Mechanismen erblicher Hauterkrankungen hat eine viel frühere Diagnose auf der Grundlage genetischer Techniken ermöglicht.102,162-164 Fötale DNA wird durch Amniozentese zwischen den Wochen 15 und 20 oder durch Chorionzottenentnahme zwischen den Wochen 10 und 12 erhalten. Das Risiko eines fetalen Verlustes mit diesen Techniken ist geringer als zwischen 0,5% und 1%.165 Weitere nichtinvasive Methoden in der Entwicklung sind die Analyse von fötaler Zell-DNA und freier fötaler DNA im mütterlichen kreislauf166 sowie die Verwendung von 3-dimensionalem Ultraschall.167,168
Die genetische Präimplantationsdiagnostik könnte auch in In-vitro-Fertilisationstechniken möglich sein, so dass nur befruchtete Eizellen, die frei von der Mutation sind, in die Gebärmutter implantiert werden, wodurch in den meisten Fällen eine Abtreibung vermieden wird.169
Zukünftige Strategien zur genetischen Behandlung der Ichthyose
Obwohl bei der genetischen Diagnose der Ichthyose wichtige Fortschritte erzielt wurden, werden auch für diese Krankheiten neue Strategien verfolgt.170 Die Haut ist das am besten zugängliche Organ für Gentransfertherapien, und daher sind solche Techniken minimal invasiv.171 Die Haut weist jedoch auch einzigartige immunologische Eigenschaften auf, die die langfristige Expression eines transgenen Produkts nicht begünstigen.172 In LI gelang es einem Prozess des Ex-vivo-Gentransfers, die normale TGM1-Expression wiederherzustellen und den Phänotyp der auf den Rücken immunsupprimierter Mäuse transplantierten Haut zu korrigieren.173.174 Kürzlich wurde auch der Phänotyp kultivierter Keratinozyten von Patienten mit HI aufgrund von Mutationen im ABCA12-Gen gewonnen.3
Interessenkonflikte
Die Autoren erklären, dass sie keine Interessenkonflikte haben.