Die Genetik der Lippen- und Gaumenspalte
 Intro / Abstractdie Lippen- und Gaumenspalte mit oder ohne Gaumenspalte ist eine komplexe angeborene Anomalie, die isoliert oder zusammen mit anderen Missbildungen beobachtet werden kann. Es kann auch Teil des Phänotyps eines genetischen Syndroms sein. Dieser Artikel dient als Übersicht über die Prävalenz von Lippen- und Gaumenspalten, das Risiko eines erneuten Auftretens und das Risiko für andere angeborene Anomalien. Genetische Syndrome und teratogene Expositionen, von denen bekannt ist, dass sie mit oralen Spalten assoziiert sind, werden untersucht. In Ergänzung, Gentests, die häufig in der pädiatrischen klinischen Genetik für die Beurteilung des Patienten mit Lippen- und Gaumenspalten werden diskutiert.
Intro / Abstractdie Lippen- und Gaumenspalte mit oder ohne Gaumenspalte ist eine komplexe angeborene Anomalie, die isoliert oder zusammen mit anderen Missbildungen beobachtet werden kann. Es kann auch Teil des Phänotyps eines genetischen Syndroms sein. Dieser Artikel dient als Übersicht über die Prävalenz von Lippen- und Gaumenspalten, das Risiko eines erneuten Auftretens und das Risiko für andere angeborene Anomalien. Genetische Syndrome und teratogene Expositionen, von denen bekannt ist, dass sie mit oralen Spalten assoziiert sind, werden untersucht. In Ergänzung, Gentests, die häufig in der pädiatrischen klinischen Genetik für die Beurteilung des Patienten mit Lippen- und Gaumenspalten werden diskutiert.
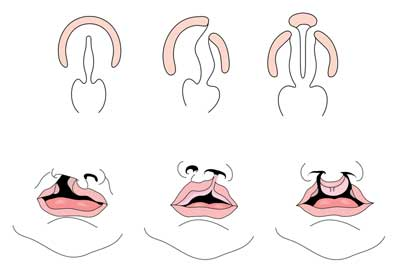
Lippenspalte mit oder ohne Gaumenspalte (CL / CP) unterscheidet sich von einer isolierten Gaumenspalte (CP) auf embryonaler, epidemiologischer und genetischer Ebene. Die Lippenspalte resultiert typischerweise daraus, dass der Oberkiefervorsprung und der mediale Nasenvorsprung zwischen der fünften und sechsten Woche der Embryonalentwicklung nicht verschmelzen. Die normale Gaumenentwicklung resultiert aus der Bildung des primären Gaumens und des sekundären Gaumens. Der primäre Gaumen wird in den Wochen sechs bis sieben durch die Entwicklung und Fusion der medialen Nasen-, lateralen Nasen- und Oberkieferfortsätze gebildet. Der sekundäre Gaumen stammt aus den Gaumenregalen (die sich aus den gepaarten Oberkieferfortsätzen des ersten Zweigbogens entwickeln), die horizontal werden und verschmelzen und etwa in der neunten Woche der Embryonalentwicklung den harten und weichen Gaumen bilden. Regale verschmelzen auch mit dem primären Gaumen und der Nasenscheidewand. (1)
Mundspalten sind mit einer Gesamtprävalenz von 1 einer der häufigsten Geburtsfehler im Neugeborenenalter.6 pro tausend Neugeborene weltweit, mit CL / CP in etwa einem pro tausend Geburten und CP in 0,6 pro tausend Geburten gesehen. (2) Es gibt eine höhere Häufigkeit von CL / CP bei Personen asiatischer, afrikanischer und indianischer Abstammung. CL / CP ist auch häufiger bei Männern. Im Gegensatz, Es gibt keinen signifikanten Unterschied in der Inzidenz von CP zwischen verschiedenen ethnischen Hintergründen, und CP tritt häufiger bei Frauen auf. (3) Das Risiko eines erneuten Auftretens innerhalb einer Familie hängt davon ab, ob die Spalte isoliert (ohne andere klinische Befunde) oder als Teil eines genetischen Syndroms angesehen wird. Die meisten Fälle von oralen Spalten sind isoliert (ungefähr 80%). Es wird angenommen, dass isolierte Spalten eine multifaktorielle Vererbung aufweisen: Sie sind auf eine Kombination mehrerer genetischer und umweltbedingter Faktoren zurückzuführen. Das Rezidivrisiko (Tabelle 1) steigt, wenn mehr als ein betroffener Angehöriger vorliegt. Das Risiko eines erneuten Auftretens steigt auch, je schwerwiegender der Defekt ist.

Lippen- und Gaumenspalte können mit anderen angeborenen Anomalien gesehen werden. Die Wahrscheinlichkeit einer genetischen oder teratogenen Ätiologie steigt, je mehr angeborene Anomalien ein Patient aufweist. Das Vorhandensein anderer Probleme wie geistiger Behinderung, Verhaltensprobleme wie Autismus, dysmorphe Merkmale oder andere medizinische Bedenken machen auch eine genetische Störung oder eine teratogene Exposition wahrscheinlicher. Ungefähr 13% der Personen mit Lippenspalte haben andere medizinische Bedenken oder Anomalien. Die Zahl steigt auf 37% mit Lippen- und Gaumenspalte und auf 47% mit Gaumenspalte allein.
Es wurde gezeigt, dass die pränatale Exposition gegenüber teratogenen Wirkstoffen (wie Thalidomid, Antikonvulsiva, Alkohol, Retinsäure und Zigaretten) und Erkrankungen der Mutter (wie Diabetes, Röteln und Folatmangel) das Risiko für orale Spalten erhöht. Das Vorhandensein von Fruchtwasserbändern erhöht auch das Risiko von Spalten. Es ist bekannt, dass eine perikonzeptuelle Folsäure-Supplementierung das Risiko von oralen Spalten verringert.
Die Pierre-Robin-Sequenz ist eine kraniofaziale Anomalie, die durch Hypoplasie oder Mikrognathie des Unterkiefers, sekundäre U-förmige Gaumenspalte und Glossoptose gekennzeichnet ist, die zu obstruktiver Apnoe und Fütterungsschwierigkeiten führt. Die Pierre-Robin-Sequenz kann als Teil genetischer Syndrome angesehen werden (22q11.2-Deletionssyndrom, Stickler-Syndrom; unten beschrieben). (5)
Es gibt Hunderte von genetischen Syndromen, die mit oralen Spalten assoziiert sind, einschließlich zytogenetischer Anomalien (Aneuploidien, Mikrodeletionen) und Einzelgen-Störungen (Mendelsche Störungen). Die Bestätigung einer genetischen Diagnose ist wichtig, um die Prognose zu bestimmen und ein Rezidivrisiko festzustellen.
Aneuploidien wie Trisomie 13 und 18 haben eine starke Assoziation mit CL/CP. Trisomie 13 (auch bekannt als Patau-Syndrom) ist mit drei Kopien von Chromosom 13 oder unausgeglichenen Robertsschen Translokationen mit Chromosom 13 assoziiert. Babys, die mit dieser Erkrankung geboren wurden, sterben normalerweise in der Neugeborenenperiode. Zu den klinischen Merkmalen gehören Lippen- und Gaumenspalte, Wachstumsverzögerung, schwere Fehlbildungen des Zentralnervensystems (einschließlich Holoprosencephalie), Mikrozephalie, Mikrophthalmie, Iriskolobom, Fehlen der Augen, Fehlbildungen der Ohren, Polydaktylie, geballte Fäuste, Rocker-Bottom-Füße, angeborene Herzfehler und Urogenitaldefekte. Mittellinienspalten (ansonsten sehr selten) können bei Trisomie 13 aufgrund des Risikos für Mittelliniendefekte, einschließlich Holoprosencephalie, beobachtet werden. Trisomie 18 (auch bekannt als Edwards-Syndrom) ist typischerweise auf drei verschiedene Kopien von Chromosom 18 zurückzuführen und ist mit einem schlechten postnatalen Ergebnis verbunden. Klinische Merkmale sind Lippen- und Gaumenspalte, geistige Behinderung, Gedeihstörungen, angeborene Herzkrankheit, Hypertonie, Mikrognathie, kurzes Brustbein, niedrig gesetzte missgebildete Ohren, geballte Hände, Rocker-Bottom-Füße und hypoplastische Nägel, unter anderem. Die Trisomie 13 und 18 kann durch Chromosomenanalyse (Karyotypisierung) leicht bestätigt oder ausgeschlossen werden.
Mikrodeletionssyndrome beinhalten typischerweise die Deletion eines Teils eines Chromosoms. Diese Deletionen sind möglicherweise zu klein, um durch Standard-Karyotypisierung nachgewiesen zu werden, und erfordern möglicherweise den Nachweis von FISH (Fluoreszenz-in-situ-Hybridisierung) oder Microarray-Technologie. Ein bekanntes Mikrodeletionssyndrom im Zusammenhang mit Gaumenspalten ist das 22q11.2-Deletionssyndrom (auch bekannt als Digeorge / Velocardiofacial-Syndrom). Gaumenanomalien einschließlich velopharyngealer Inkompetenz, submuköse Spalten, bifide Uvula und Gaumenspalte treten bei 69% der Personen mit 22q11.2-Deletion auf und können Teil der Pierre-Robin-Sequenz sein. Andere klinische Befunde umfassen angeborene Herzfehler, Hörverlust, dysmorphe Merkmale, Immunschwäche, Hypokalzämie, Nierenanomalien, Ernährungsprobleme, Skelettanomalien und psychiatrische Störungen. Es wird angenommen, dass etwa 10% der Fälle des 22q11.2-Deletionssyndroms familiär sind. Die Deletion segregiert autosomal dominant.(6) Das Wolf-Hirschhorn-Syndrom, das auf eine Deletion im kurzen Arm von Chromosom 4 zurückzuführen ist, ist auch mit oralen Spalten assoziiert (bei 25% bis 50% der betroffenen Personen). Charakteristische Gesichtszüge (einschließlich prominenter Glabella, die zu einem “griechisch-kriegerischen Helm-Aussehen” führen), angeborene Herzfehler, geistige Behinderung, Krampfanfälle, Gedeihstörungen, Mikrognathie, präaurikuläre Markierungen oder Gruben und Hypodontie können ebenfalls als Teil der Erkrankung angesehen werden.(7)
Einzelgenstörungen mit oralen Spalten umfassen unter anderem das Stickler-Syndrom, das Treacher-Collins-Syndrom und das Van-der-Woude-Syndrom. Das Stickler-Syndrom ist eine Kollagenerkrankung mit autosomal dominanter und seltener autosomal rezessiver Vererbung. Gemeinsame Merkmale sind Gaumenspalte (gesehen als Teil der Pierre-Robin-Sequenz oder ohne Mikrognathie), Hörverlust (sensorineural und leitend), Skelettbefunde (früh einsetzende Arthritis, spondyloepiphysäre Dysplasie), Augenanomalien (hohe Myopie, Glaskörperanomalien) und charakteristische Gesichtszüge (mit Unterentwicklung des Oberkiefers und der Nasenbrücke, Mittelgesichtsretrusion). Gentests für das Stickler-Syndrom können komplex sein, da Mutationen in mindestens sechs Genen bei betroffenen Personen beschrieben wurden. Etwa 90% der Patienten mit Stickler-Syndrom haben Mutationen im COL2A1-Gen und eine autosomal dominante Form der Erkrankung.(8) Das Treacher-Collins-Syndrom ist eine autosomal dominante Erkrankung, die bei 28% der Betroffenen durch Gaumenspalte mit oder ohne Lippenspalte gekennzeichnet ist. Andere Anomalien umfassen Hypoplasie der Jochbeinknochen und des Unterkiefers, Anomalien des äußeren Ohrs, Kolobom des unteren Augenlids, Schallleitungsschwerhörigkeit, Fehlen der unteren Wimpern, präaurikuläre Haarverlagerung auf die Wangen und Choanalstenose oder Atresie. Die Diagnose des Treacher-Collins-Syndroms basiert auf klinischen und radiologischen Befunden. Mutationen in mindestens drei Genen wurden beschrieben, wobei Mutationen in TCOF1 bei 78% bis 93% der Patienten beobachtet wurden.(9) Das Van-der-Woude-Syndrom ist gekennzeichnet durch angeborene, meist bilaterale paramedische Unterlippenfisteln (Gruben) oder manchmal kleine Hügel mit einem Sinustrakt, der von einer Schleimdrüse der Lippe führt, und Mundspalten (einschließlich CL / CP und CP). Van der Woude ist eine autosomal dominante Erkrankung, die mit Mutationen im IRF6-Gen assoziiert ist (10). Das Testen auf Einzelgen- oder Multigenbedingungen erfordert eine direkte Analyse des Gens durch Sequenzierung und / oder Deletions- / Duplikationsanalyse (wie MLPA).
Da genetische Syndrome mit Lippen- und Gaumenspalten mit Aneuploidien, Chromosomenmikrodeletionen / Mikroduplikationen oder Einzelgenstörungen in Verbindung gebracht werden können, können Gentests ein komplizierter Prozess sein. Eine gründliche Anamnese, ein Stammbaum aus drei Generationen, eine Schwangerschaftsgeschichte und eine dysmorphologische Untersuchung durch einen klinischen Genetiker können das klinische Bild klären und gezielte Gentests ermöglichen. Neuere Technologien, einschließlich Microarray, ermöglichen die Identifizierung kleiner Mikrodeletionen und Mikroduplikationen, die zuvor bei der Standard-Karyotypisierung übersehen wurden. Leider führt diese Technik auch zur Identifizierung von Deletionen und Duplikationen unbekannter klinischer Bedeutung, was den genetischen Beratungsprozess erschwert. Das Testen auf Einzelgenstörungen oder Mendelsche Störungen erfordert die klinische Verfügbarkeit von Gentests für das gewünschte Gen. Es kann auch teuer sein, wenn es nicht von der Krankenversicherung abgedeckt wird. Neue Technologien wie Next-Generation-Sequenzierung, Exomsequenzierung oder Genomsequenzierung (zusammen als genomische Tests bezeichnet) sind jetzt klinisch verfügbar. Durch die gleichzeitige Analyse von Hunderten bis Tausenden von Genen erhöhen diese Tests die diagnostische Leistungsfähigkeit und den Ertrag erheblich. Im Vergleich zu anderen Techniken können diese Tests schneller und kostengünstiger eine Antwort liefern. Im Forschungsbereich hat die Exom- und Genomsequenzierung zur Identifizierung neuer Gene sowie zur Erweiterung der klinischen Merkmale und des Spektrums für genetische Mutationen geführt. Wie bei der Microarray-Technologie können genomische Tests Syndrome erkennen, die nicht mit der Präsentation des Patienten und / oder dem Testgrund zusammenhängen. Angesichts der inhärenten Komplexität von Gentests ist eine Einverständniserklärung erforderlich.
Schlussfolgerung
Obwohl die Lippen- und Gaumenspalte in den meisten Fällen eine isolierte Anomalie darstellt, besteht ein starker Zusammenhang zwischen Mundspalten und anderen Anomalien und genetischen Syndromen. Eine genetische Bewertung durch einen klinischen Genetiker und einen genetischen Berater ist für eine vorausschauende Anleitung und zur Bestimmung des Rezidivrisikos unerlässlich. Gentests, die eine Einverständniserklärung erfordern, können während einer genetischen Bewertung koordiniert und interpretiert werden.
Anya Revah, MS, ist die leitende genetische Beraterin in der Abteilung für medizinische Genetik am Maimonides Infants and Children’s Hospital in Brooklyn, New York. Sie ist auch aktives Mitglied des multidisziplinären Teams des Maimonides Medical Center und des Kings County Hospital Cleft Lip and Palate. Sie hat einen Master in Science in genetischer Beratung von der Boston University in Boston, Massachusetts.
1. In: Sadler TW. Langmans medizinische Embryologie. Neunte Auflage. Seiten 390-395.
2. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. Für das National Birth Defects Prevention Network. Aktualisierte Schätzungen der nationalen Geburtenprävalenz für ausgewählte Geburtsfehler in den Vereinigten Staaten. 2004-2006. Geburtsfehlerforschung (Teil A): Klinische und molekulare Teratologie 2010; 88: 1008-1016.
3. Fraser FC. Die Genetik der Lippen- und Gaumenspalte. Uhr. J. Brummen. Genet. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC, et al. Perikonzeptuelle Folataufnahme durch Ergänzung und Nahrungsaufnahme reduziert das Risiko einer nicht-syndromischen Lippenspalte mit oder ohne Gaumenspalte. Zurück Med 2004;39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Entwicklungs- und genetische Perspektiven auf Pierre Robin Sequenz. Uhr. Dr. Med. Genet. 2013;163C:295-305.
6. McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11.2 Deletionssyndrom. Sept. 23, 1999. . In: Köhler, H., et al., Herausgeberanmerkung. GeneReviews . Seattle (WA): Universität von Washington, Seattle; 1993-2014. Verfügbar ab: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia A, Carey JC, Süd-ST, et al. Wolf-Hirschhorn-Syndrom. Apr. 29, 2002. . In: Köhler, H., et al., Herausgeberanmerkung. GeneReviews . Seattle (WA): Universität von Washington, Seattle; 1993-2014. Verfügbar ab: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, Ala-Kokko L. Stickler-Syndrom. Jun. 9, 2000. . In: Köhler, H., et al., Herausgeberanmerkung. GeneReviews . Seattle (WA): Universität von Washington, Seattle; 1993-2014. Verfügbar ab: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs EW. Treacher-Collins-Syndrom. Jul. 20, 2004. . In: Köhler, H., et al., Herausgeberanmerkung. GeneReviews . Seattle (WA): Universität von Washington, Seattle; 1993-2014. Verfügbar ab: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte B.C., Saal H.M., Goudy S., et al. IRF6-bedingte Störungen. Oct. 30, 2003. . In: Köhler, H., et al., Herausgeberanmerkung. GeneReviews . Seattle (WA): Universität von Washington, Seattle; 1993-2014. Verfügbar ab: http://www.ncbi.nlm.nih.gov/books/NBK1407/.